What is pseduobulk?
Many of you have heard about bulk-RNAseq data. What is pseduobulk?
Single-cell RNAseq can profile the gene expression at single-cell resolution. For differential expression, psedobulk seems to perform really well(see paper muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data). To create a pseudobulk, one can artificially add up the counts for cells from the same cell type of the same sample.
In this blog post, I’ll guide you through the art of creating pseudobulk data from scRNA-seq experiments. By the end, you’ll have the skills to transform complex single-cell data into manageable, meaningful results, and learn skills to explore and make sense of the results.
Download the data
The data are from this paper A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells
GEO accession: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154763
Get the data from ftp https://ftp.ncbi.nlm.nih.gov/geo/series/GSE154nnn/GSE154763/suppl/
On command line
wget -nH --cut-dirs=5 --no-clobber --convert-links --random-wait -r -p -E -e robots=off https://ftp.ncbi.nlm.nih.gov/geo/series/GSE154nnn/GSE154763/suppl/
ls *gz | xargs gunzipuse https://github.com/immunitastx/recover-counts to convert the log normalized the counts back to raw counts.
## for one file
./recover_counts_from_log_normalized_data.py -m 10000 -d CSV GSE154763_ESCA_normalized_expression.csv -o GSE154763_ESCA_raw_counts.txt
## for all files
# mamba install parallel
# regular expression to remove the normalized_expression.csv and rename the output to _raw_counts.txt
ls *expression.csv | parallel --rpl '{%(.+?)} s/$$1$//;' ./recover_counts_from_log_normalized_data.py -m 10000 -d CSV {} -o {%_normalized_expression.csv}_raw_count.txt
Switch to R
load libraries
library(here)
library(stringr)
library(dplyr)
library(ggplot2)
library(Seurat)
library(purrr)
library(readr)
library(harmony)
library(scCustomize)
library(SeuratDisk)
library(ggpointdensity)
library(viridis)
library(grid)read in counts files
## the count matrix is cell x gene
read_counts<- function(file){
x<- read_csv(file)
x<- as.data.frame(x)
cells<- x$index
mat<- as.matrix(x[,-1])
rownames(mat)<- cells
mat<- t(mat)
return(mat)
}
counts_files<- list.files("~/blog_data/GSE154763", full.names = TRUE, pattern = "*raw_count.txt")
counts_files#> [1] "/Users/tommytang/blog_data/GSE154763/GSE154763_cDC2_raw_count.txt"
#> [2] "/Users/tommytang/blog_data/GSE154763/GSE154763_ESCA_raw_count.txt"
#> [3] "/Users/tommytang/blog_data/GSE154763/GSE154763_KIDNEY_raw_count.txt"
#> [4] "/Users/tommytang/blog_data/GSE154763/GSE154763_LYM_raw_count.txt"
#> [5] "/Users/tommytang/blog_data/GSE154763/GSE154763_MYE_raw_count.txt"
#> [6] "/Users/tommytang/blog_data/GSE154763/GSE154763_OV-FTC_raw_count.txt"
#> [7] "/Users/tommytang/blog_data/GSE154763/GSE154763_PAAD_raw_count.txt"
#> [8] "/Users/tommytang/blog_data/GSE154763/GSE154763_THCA_raw_count.txt"
#> [9] "/Users/tommytang/blog_data/GSE154763/GSE154763_UCEC_raw_count.txt"# only use the last 3 cancer types to demonstrate, as the data are too big.
counts_files<- counts_files %>%
tail(n=3)
samples<- map_chr(counts_files, basename)
samples<- str_replace(samples, "(GSE[0-9]+)_(.+)_raw_count.txt", "\\2")
names(counts_files)<- samples
counts<- purrr::map(counts_files, read_counts)read in metadata for each cancer
## the tissue column contain some non-tumor cells
read_meta<- function(file){
y<- read_csv(file, guess_max = 100000,
col_types = cols(tissue = col_character()))
y<- as.data.frame(y)
cells<- y$index
y<- y[,-1]
rownames(y)<- cells
return(y)
}
meta_files<- list.files("~/blog_data/GSE154763", full.names = TRUE, pattern = "metadata.csv")
# take only 3 cancer types
meta_files<- meta_files %>% tail(n=3)
meta_names<- map_chr(meta_files, basename)
meta_names<- str_replace(meta_names, "(GSE[0-9]+)_(.+)_metadata.csv", "\\2")
names(meta_files)<- meta_names
meta<- purrr::map(meta_files, read_meta)Different cancer types have different number of genes.
map(counts, nrow)#> $PAAD
#> [1] 14140
#>
#> $THCA
#> [1] 15211
#>
#> $UCEC
#> [1] 15849How many genes are in common among the 3 datasets?
map(counts, ~rownames(.x)) %>%
purrr::reduce( function(x,y) {intersect(x,y)}) %>%
length()#> [1] 13762make Seurat objects
objs<- purrr::map2(counts, meta,
~ CreateSeuratObject(counts = as(.x, "sparseMatrix"),
meta.data = .y))
## remain only the tumor samples with tissue == T
#subset(objs$ESCA, subset = tissue == "T")
objs<- purrr::map(objs, ~ subset(.x, subset = tissue == "T"))
objs#> $PAAD
#> An object of class Seurat
#> 14140 features across 2628 samples within 1 assay
#> Active assay: RNA (14140 features, 0 variable features)
#>
#> $THCA
#> An object of class Seurat
#> 15211 features across 4171 samples within 1 assay
#> Active assay: RNA (15211 features, 0 variable features)
#>
#> $UCEC
#> An object of class Seurat
#> 15849 features across 4724 samples within 1 assay
#> Active assay: RNA (15849 features, 0 variable features)It is a list of 3 seurat objects.
preprocessSeurat<- function(obj){
obj<-
NormalizeData(obj, normalization.method = "LogNormalize", scale.factor = 10000) %>%
FindVariableFeatures( selection.method = "vst", nfeatures = 2000) %>%
ScaleData() %>%
RunPCA() %>%
RunHarmony(group.by.vars = "patient", dims.use = 1:30) %>%
RunUMAP(reduction = "harmony", dims = 1:30) %>%
FindNeighbors(reduction = "harmony", dims = 1:30) %>%
FindClusters(resolution = 0.6)
return(obj)
}
objs<- purrr::map(objs, preprocessSeurat)#> Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
#>
#> Number of nodes: 2628
#> Number of edges: 99201
#>
#> Running Louvain algorithm...
#> Maximum modularity in 10 random starts: 0.8476
#> Number of communities: 10
#> Elapsed time: 0 seconds
#> Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
#>
#> Number of nodes: 4171
#> Number of edges: 167212
#>
#> Running Louvain algorithm...
#> Maximum modularity in 10 random starts: 0.9003
#> Number of communities: 17
#> Elapsed time: 0 seconds
#> Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
#>
#> Number of nodes: 4724
#> Number of edges: 172629
#>
#> Running Louvain algorithm...
#> Maximum modularity in 10 random starts: 0.8709
#> Number of communities: 15
#> Elapsed time: 0 secondsp1<- scCustomize::DimPlot_scCustom(objs$UCEC)
p2<- scCustomize::DimPlot_scCustom(objs$UCEC, group.by = "MajorCluster")
p1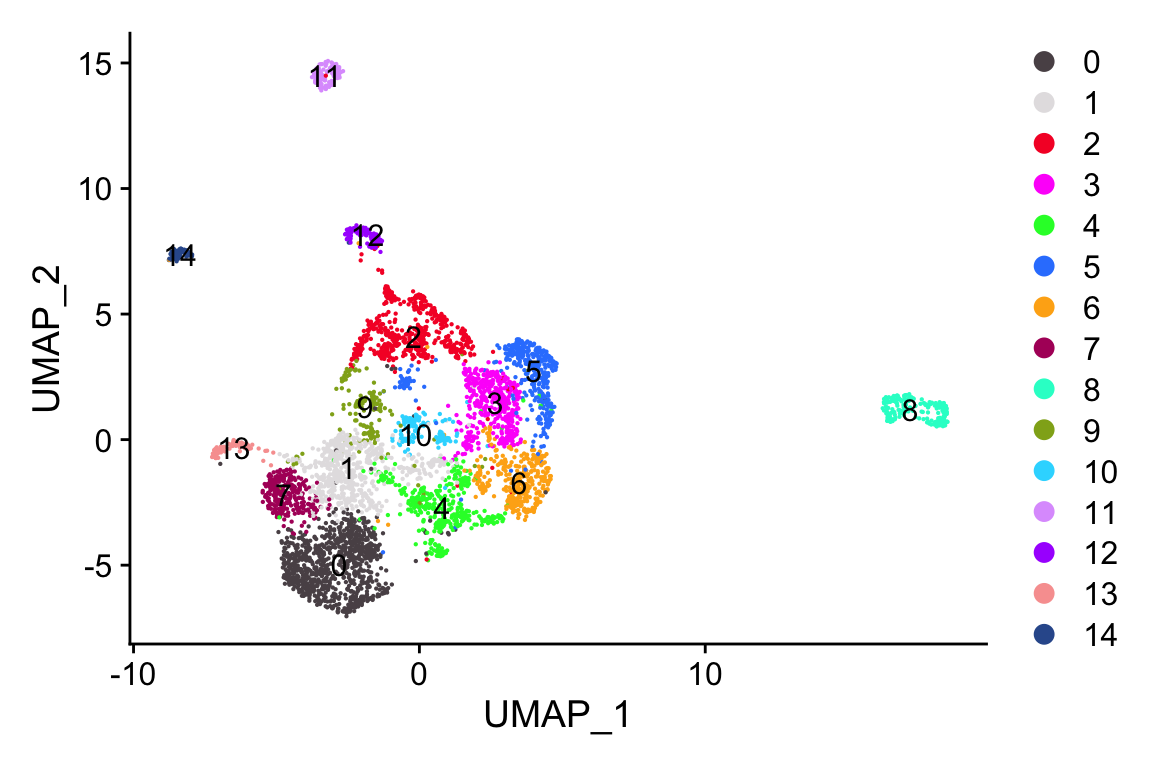
p2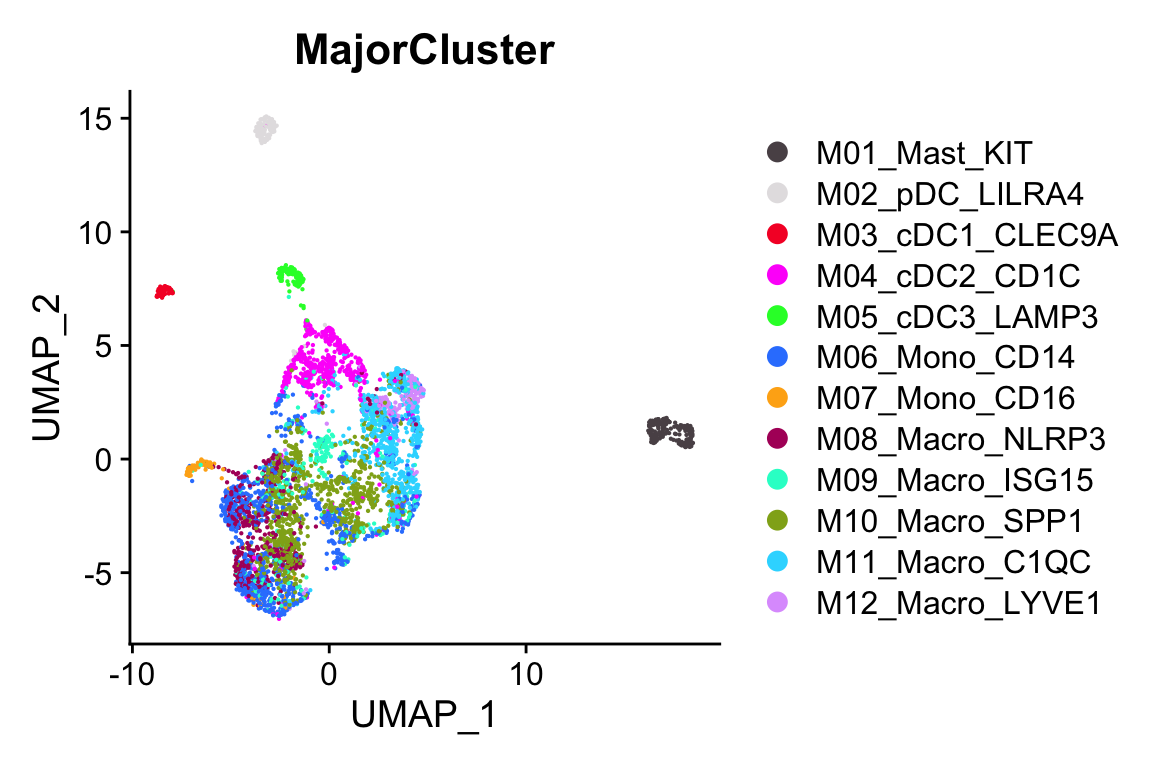
Create psedobulk for the single cell data
Clean up the cell type annotation so it is consistent across cancer types.
purrr::map(objs, ~.x$MajorCluster %>% unique() %>% sort())#> $PAAD
#> [1] "M01_Mast_KIT" "M03_cDC1_CLEC9A" "M04_cDC2_CD1C" "M05_cDC3_LAMP3"
#> [5] "M06_Mono_CD14" "M07_Mono_CD16" "M08_Macro_SPP1" "M09_Macro_C1QC"
#>
#> $THCA
#> [1] "M01_Mast_KIT" "M02_pDC_LILRA4" "M03_cDC1_CLEC9A" "M04_cDC2_CD1C"
#> [5] "M05_cDC3_LAMP3" "M06_Mono_CD14" "M07_Mono_CD16" "M08_Macro_NLRP3"
#> [9] "M09_Macro_SPP1" "M10_Macro_C1QC" "M11_Macro_ISG15" "M12_Macro_LYVE1"
#> [13] "M13_Macro_INHBA"
#>
#> $UCEC
#> [1] "M01_Mast_KIT" "M02_pDC_LILRA4" "M03_cDC1_CLEC9A" "M04_cDC2_CD1C"
#> [5] "M05_cDC3_LAMP3" "M06_Mono_CD14" "M07_Mono_CD16" "M08_Macro_NLRP3"
#> [9] "M09_Macro_ISG15" "M10_Macro_SPP1" "M11_Macro_C1QC" "M12_Macro_LYVE1"clean_cluster_name<- function(obj){
annotation<- obj@meta.data %>%
mutate(annotation = str_replace(MajorCluster, "^M[0-9]{2}_", "")) %>%
pull(annotation)
obj$annotation<- annotation
return(obj)
}
objs<- purrr::map(objs, clean_cluster_name)
purrr::map(objs, ~.x$annotation %>% unique() %>% sort())#> $PAAD
#> [1] "cDC1_CLEC9A" "cDC2_CD1C" "cDC3_LAMP3" "Macro_C1QC" "Macro_SPP1"
#> [6] "Mast_KIT" "Mono_CD14" "Mono_CD16"
#>
#> $THCA
#> [1] "cDC1_CLEC9A" "cDC2_CD1C" "cDC3_LAMP3" "Macro_C1QC" "Macro_INHBA"
#> [6] "Macro_ISG15" "Macro_LYVE1" "Macro_NLRP3" "Macro_SPP1" "Mast_KIT"
#> [11] "Mono_CD14" "Mono_CD16" "pDC_LILRA4"
#>
#> $UCEC
#> [1] "cDC1_CLEC9A" "cDC2_CD1C" "cDC3_LAMP3" "Macro_C1QC" "Macro_ISG15"
#> [6] "Macro_LYVE1" "Macro_NLRP3" "Macro_SPP1" "Mast_KIT" "Mono_CD14"
#> [11] "Mono_CD16" "pDC_LILRA4"Create Psedobulk
There are many ways to do it.
Let’s use presto for more low-level analysis instead of using the wrapper functions in muscat. To use muscat, you will need to convert the Seurat object to a SingleCellExperiment object.
library(presto)
objs$PAAD@meta.data %>%
head()#> orig.ident nCount_RNA nFeature_RNA percent_mito n_counts
#> AAACGGGGTGTGCCTG-35 SeuratProject 7626 2353 0.03575639 7635
#> AAAGATGAGCGATTCT-35 SeuratProject 9953 2396 0.02630258 9961
#> AAAGATGCAATTGCTG-35 SeuratProject 11091 2930 0.04469274 11098
#> AAAGATGCATGGTCTA-35 SeuratProject 9739 2551 0.03981121 9746
#> AAAGTAGCAAACGCGA-35 SeuratProject 6959 1145 0.02802030 9850
#> AAATGCCTCCACGACG-35 SeuratProject 2404 1031 0.03742204 2405
#> percent_hsp barcode batch library_id cancer
#> AAACGGGGTGTGCCTG-35 0.020563196 AAACGGGGTGTGCCTG 35 PACA-P20181121-T PAAD
#> AAAGATGAGCGATTCT-35 0.003312921 AAAGATGAGCGATTCT 35 PACA-P20181121-T PAAD
#> AAAGATGCAATTGCTG-35 0.004234997 AAAGATGCAATTGCTG 35 PACA-P20181121-T PAAD
#> AAAGATGCATGGTCTA-35 0.031089678 AAAGATGCATGGTCTA 35 PACA-P20181121-T PAAD
#> AAAGTAGCAAACGCGA-35 0.015837563 AAAGTAGCAAACGCGA 35 PACA-P20181121-T PAAD
#> AAATGCCTCCACGACG-35 0.007484408 AAATGCCTCCACGACG 35 PACA-P20181121-T PAAD
#> patient tissue n_genes MajorCluster source tech
#> AAACGGGGTGTGCCTG-35 P20181121 T 2362 M01_Mast_KIT ZhangLab 10X5
#> AAAGATGAGCGATTCT-35 P20181121 T 2404 M06_Mono_CD14 ZhangLab 10X5
#> AAAGATGCAATTGCTG-35 P20181121 T 2937 M08_Macro_SPP1 ZhangLab 10X5
#> AAAGATGCATGGTCTA-35 P20181121 T 2558 M07_Mono_CD16 ZhangLab 10X5
#> AAAGTAGCAAACGCGA-35 P20181121 T 2891 M01_Mast_KIT ZhangLab 10X5
#> AAATGCCTCCACGACG-35 P20181121 T 1032 M01_Mast_KIT ZhangLab 10X5
#> UMAP1 UMAP2 RNA_snn_res.0.6 seurat_clusters
#> AAACGGGGTGTGCCTG-35 7.324396 -0.6997318 4 4
#> AAAGATGAGCGATTCT-35 -5.009157 -0.8700839 2 2
#> AAAGATGCAATTGCTG-35 -5.944770 1.9433328 7 7
#> AAAGATGCATGGTCTA-35 -3.173428 -2.7142875 2 2
#> AAAGTAGCAAACGCGA-35 7.285788 -0.4640464 4 4
#> AAATGCCTCCACGACG-35 4.864260 -0.9610466 4 4
#> annotation
#> AAACGGGGTGTGCCTG-35 Mast_KIT
#> AAAGATGAGCGATTCT-35 Mono_CD14
#> AAAGATGCAATTGCTG-35 Macro_SPP1
#> AAAGATGCATGGTCTA-35 Mono_CD16
#> AAAGTAGCAAACGCGA-35 Mast_KIT
#> AAATGCCTCCACGACG-35 Mast_KITWe will need to create a pseduobulk per cancer type, per sample, and per cell type. those are in the metadata column: ‘annotation’, ‘patient’, ‘cancer’.
create_pseduo_bulk<- function(obj){
data_collapsed <- presto::collapse_counts(obj@assays$RNA@counts,
obj@meta.data,
c('annotation', 'patient', 'cancer'))
meta_data<- data_collapsed$meta_data
mat<- data_collapsed$counts_mat
colnames(mat)<- paste(meta_data$cancer, colnames(mat), sep="_")
return(list(mat = mat, meta_data = meta_data))
}
pseduo_bulk_objs<- purrr::map(objs, create_pseduo_bulk)
### different number of genes per dataset
purrr::map(pseduo_bulk_objs, ~nrow(.x$mat)) #> $PAAD
#> [1] 14140
#>
#> $THCA
#> [1] 15211
#>
#> $UCEC
#> [1] 15849genes_per_data<- purrr::map(pseduo_bulk_objs, ~rownames(.x$mat))
## common number of genes
common_genes<- purrr::reduce(genes_per_data, intersect)
length(common_genes)#> [1] 13762subset_common_mat<- function(mat){
mat<- mat[common_genes, ]
return(mat)
}
mats<- map(pseduo_bulk_objs, ~subset_common_mat(.x$mat))
map(mats, dim)#> $PAAD
#> [1] 13762 42
#>
#> $THCA
#> [1] 13762 120
#>
#> $UCEC
#> [1] 13762 93meta_data<- map(pseduo_bulk_objs, ~.x$meta_data)
meta_data$PAAD %>% head()#> annotation patient cancer
#> 1 Mast_KIT P20181121 PAAD
#> 2 Mono_CD14 P20181121 PAAD
#> 3 Macro_SPP1 P20181121 PAAD
#> 4 Mono_CD16 P20181121 PAAD
#> 5 cDC2_CD1C P20181121 PAAD
#> 6 Macro_C1QC P20181121 PAADmats$PAAD[1:5, 1:6]#> PAAD_sample_5 PAAD_sample_6 PAAD_sample_4 PAAD_sample_7
#> FO538757.2 69 19 50 5
#> AP006222.2 0 1 0 0
#> RP4-669L17.10 0 1 0 0
#> RP11-206L10.9 0 0 2 0
#> LINC00115 2 5 6 1
#> PAAD_sample_1 PAAD_sample_3
#> FO538757.2 2 17
#> AP006222.2 0 0
#> RP4-669L17.10 0 0
#> RP11-206L10.9 0 1
#> LINC00115 0 3PCA analysis
merge all the count table into a big one (I am going to do PCA!)
mats<- purrr::reduce(mats, cbind)
meta_data<- purrr::reduce(meta_data, rbind)
dim(mats)#> [1] 13762 255final_meta<- meta_data
## log normalize the count matrix
total_reads<- colSums(mats)
final_mat<- t(t(mats)/total_reads)
final_mat<- log2(final_mat + 1)library(genefilter)
# choose the top 1000 most variabel genes
top_genes<- genefilter::rowVars(final_mat) %>%
sort(decreasing = TRUE) %>%
names() %>%
head(1000)
# subset only the top 1000 genes
expression_mat_sub<- final_mat[top_genes, ]PCA analysis. If you are new to it, read my old blog post: PCA in action.
# calculate the PCA
pca<- prcomp(t(expression_mat_sub),center = TRUE, scale. = TRUE)
# check the order of the samples are the same.
all.equal(rownames(pca$x), colnames(expression_mat_sub))#> [1] TRUEPC1_and_PC2<- data.frame(PC1=pca$x[,1], PC2= pca$x[,2])
PC1_and_PC2<- cbind(PC1_and_PC2, final_meta)
head(PC1_and_PC2)#> PC1 PC2 annotation patient cancer
#> PAAD_sample_5 -10.131179 28.929632 Mast_KIT P20181121 PAAD
#> PAAD_sample_6 16.801059 6.981512 Mono_CD14 P20181121 PAAD
#> PAAD_sample_4 18.822229 9.311594 Macro_SPP1 P20181121 PAAD
#> PAAD_sample_7 13.646380 4.989768 Mono_CD16 P20181121 PAAD
#> PAAD_sample_1 -4.800154 2.925677 cDC2_CD1C P20181121 PAAD
#> PAAD_sample_3 13.641424 7.164102 Macro_C1QC P20181121 PAADplot PCA plot
library(ggplot2)
p1<- ggplot(PC1_and_PC2, aes(x=PC1, y=PC2)) +
geom_point(aes(color = cancer)) +
theme_bw(base_size = 14) Let’s color it by cell type
library(Polychrome)
set.seed(123)
length(unique(PC1_and_PC2$annotation))#> [1] 13mypal <- kelly.colors(14)[-1]
p2<- ggplot(PC1_and_PC2, aes(x=PC1, y=PC2)) +
geom_point(aes(color = annotation)) +
theme_bw(base_size = 14) +
scale_color_manual(values = mypal %>% unname())
p1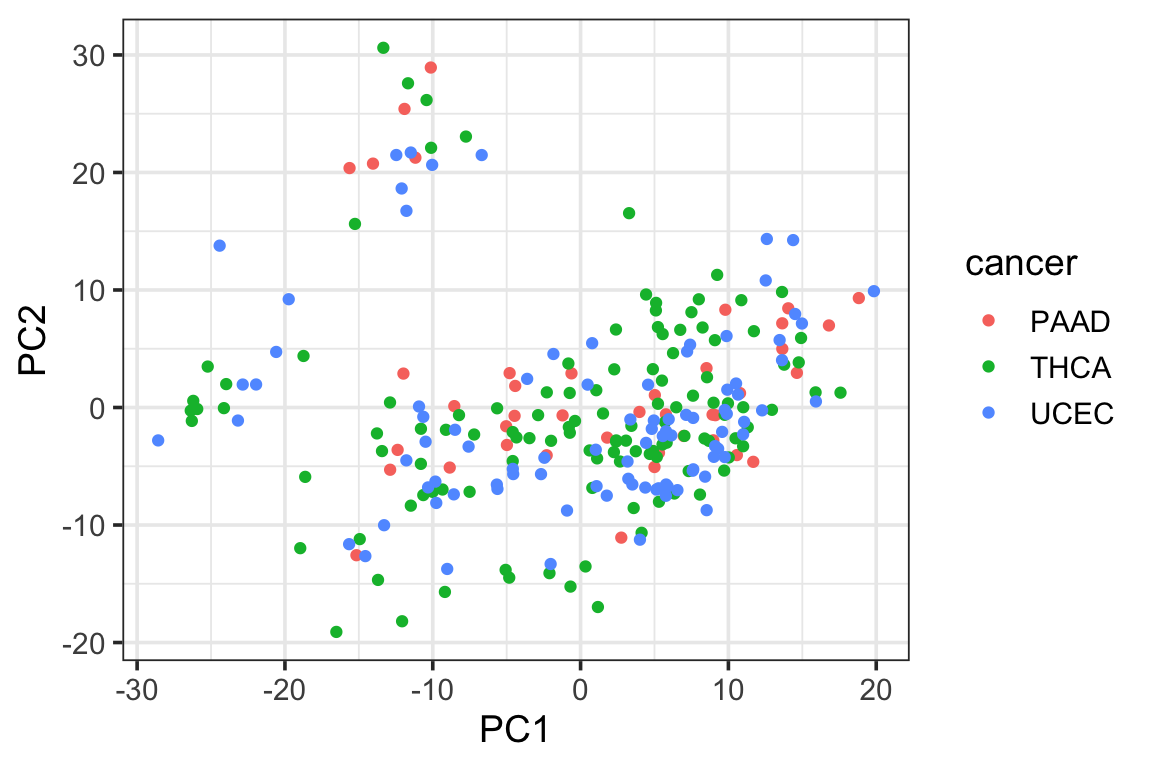
p2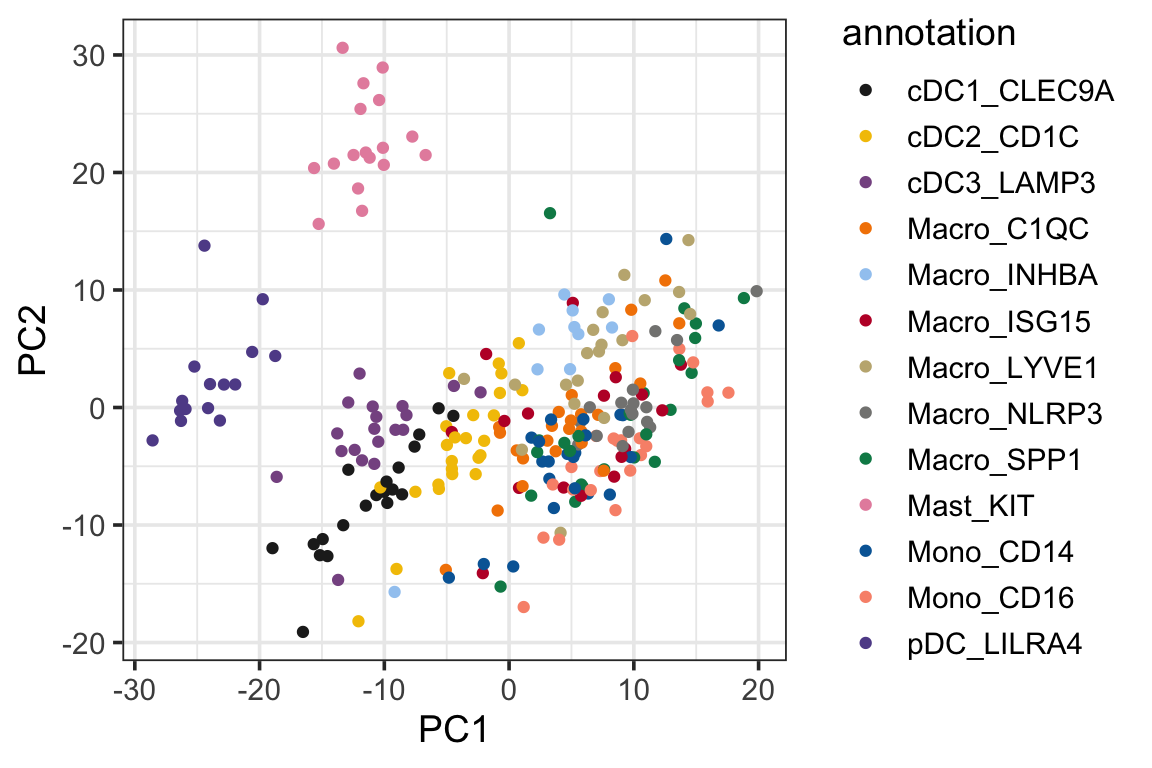
We see the psedobulk of the same myeloid cell types of different cancer types tend to cluster together which makes sense!
Visualize in heatmap.
library(RColorBrewer)
library(ComplexHeatmap)
#RColorBrewer::display.brewer.all()
set.seed(123)
cols<- RColorBrewer::brewer.pal(n = 3, name = "Set1")
unique(final_meta$cancer)#> [1] "PAAD" "THCA" "UCEC"rownames(final_meta)<- colnames(expression_mat_sub)
ha<- HeatmapAnnotation(df = final_meta[, -2],
col=list(cancer = setNames(cols, unique(final_meta$cancer) %>% sort()),
annotation= setNames(unname(mypal), unique(final_meta$annotation) %>% sort())),
show_annotation_name = TRUE)
col_fun<- circlize::colorRamp2(c(-2,0, 2), colors = c("blue", "white", "red"))
Heatmap(t(scale(t(expression_mat_sub))), top_annotation = ha,
show_row_names = FALSE, show_column_names = FALSE,
show_row_dend = FALSE,
col = col_fun,
name = "expression",
column_dend_reorder = TRUE,
raster_quality = 3,
use_raster = FALSE,
)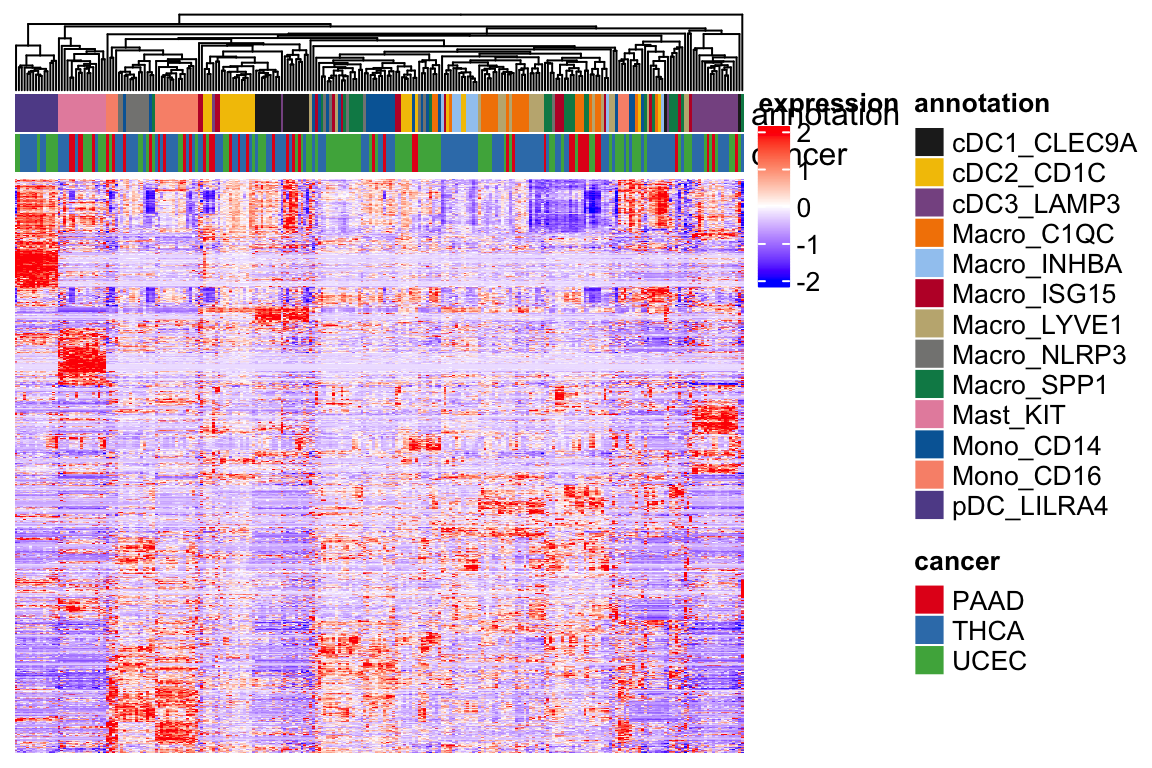
Let’s run Harmony to correct the batches of different cancer type and patients.
set.seed(123)
harmony_embeddings <- harmony::HarmonyMatrix(
expression_mat_sub, final_meta, c('cancer', 'patient'), do_pca = TRUE, verbose= TRUE
)
rownames(harmony_embeddings)<- rownames(final_meta)
harmony_pc<- data.frame(harmony1=harmony_embeddings[,1], harmony2= harmony_embeddings[,2])
harmony_pc<- cbind(harmony_pc, final_meta)
## plot PCA plot
library(ggplot2)
p1<- ggplot(harmony_pc, aes(x=harmony1, y=harmony2)) +
geom_point(aes(color = cancer)) +
theme_bw(base_size = 14) +
scale_color_manual(values = cols)
p2<- ggplot(harmony_pc, aes(x=harmony1, y=harmony2)) +
geom_point(aes(color = annotation)) +
theme_bw(base_size = 14) +
scale_color_manual(values = mypal %>% unname())
p1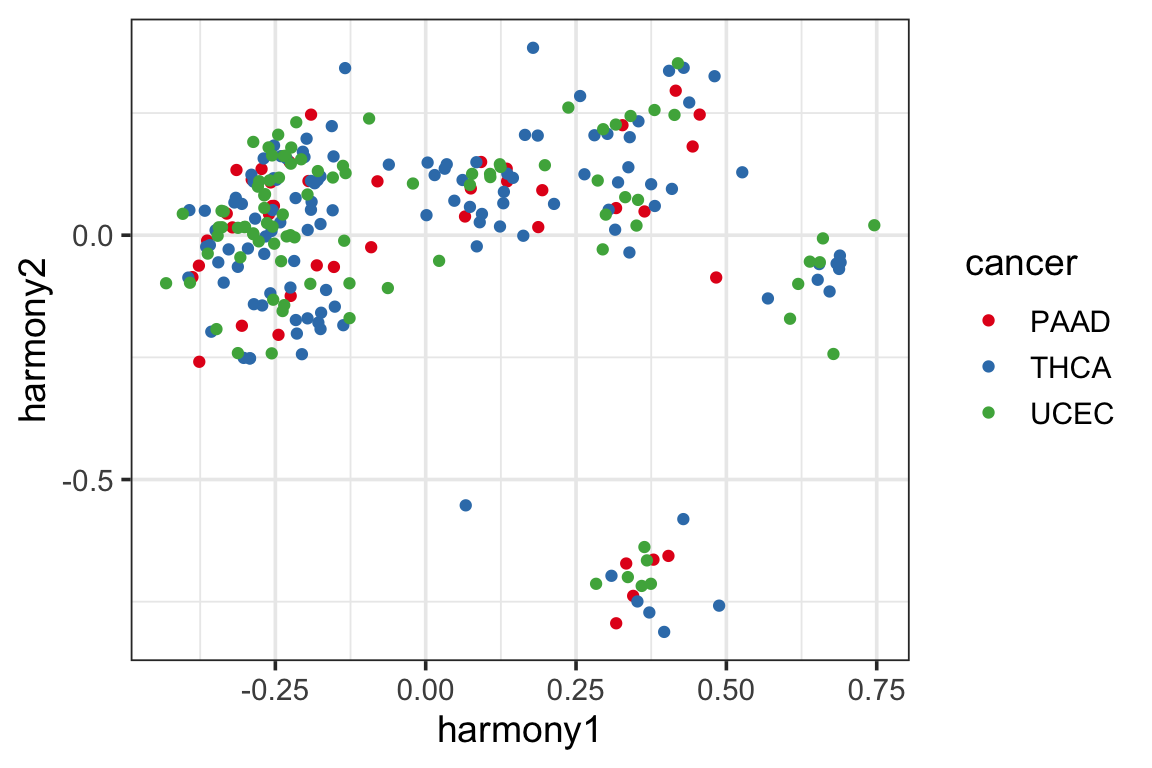
p2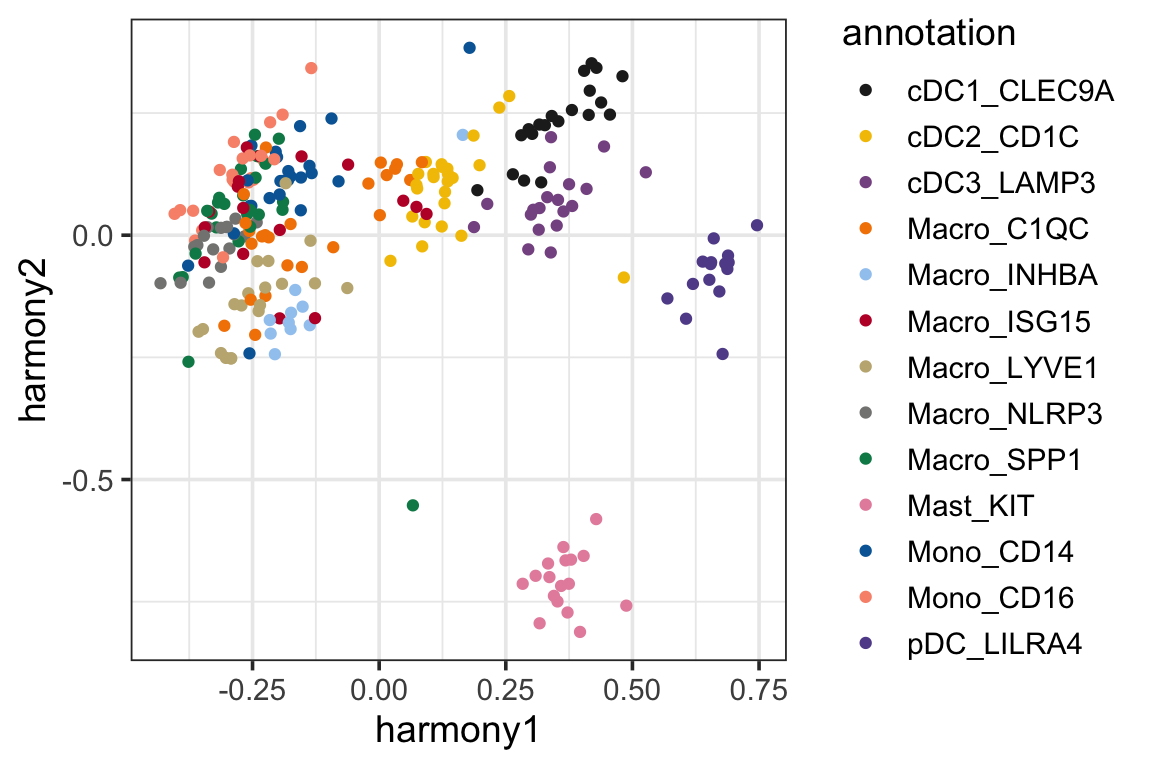
It looks like the samples are clustered a little better (which is expected as harmony artifically mixes the samples from different batches).
Read this paper Signal recovery in single cell batch integration as harmony or any single-cell integration method may erase biological differences.
What gets erased when you integrate #singlecell data across samples/studies, and can you get it back? When samples are from e.g. healthy & disease, should you simply massage cells together? FINALLY, I feel we can answer this question: https://t.co/0PrkIZDyDp pic.twitter.com/l8FbHgGrrA
— Nancy Zhang (@NancyZh60672287) September 23, 2023
Let’s visulize the heatmap by the sample correlations in the harmony space.
Heatmap(cor(t(harmony_embeddings)), top_annotation = ha, show_row_names = FALSE,
show_column_names = FALSE, column_dend_reorder = TRUE,
name="harmony space\ncorrelation",
raster_quality = 3,
use_raster = TRUE)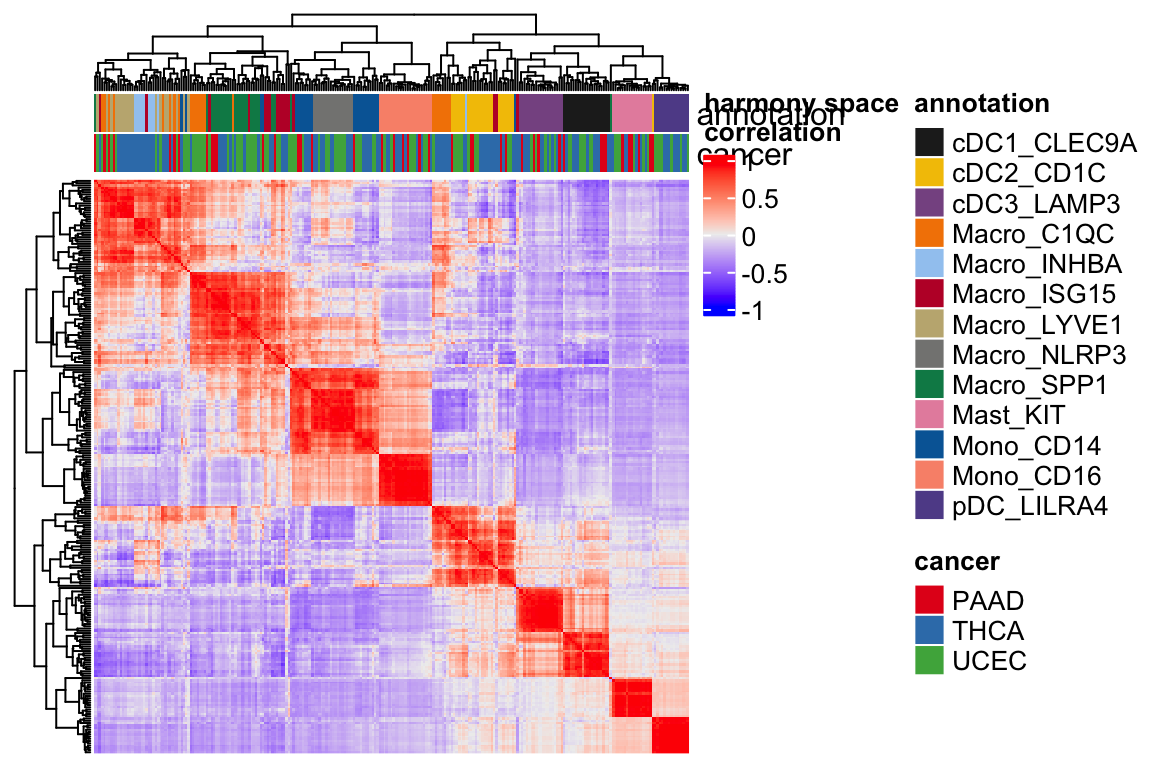 You see the annotation bar looks much cleaner.
You see the annotation bar looks much cleaner.
That’s a wrap! I hope you learned how to construct pseduobulk for single cell data.
Further reading
A blog post by Jean Fan: Create psedobulk by linear algebra
presto::collapse_counts()followed by DESeq2 workflow: https://github.com/immunogenomics/presto/blob/master/vignettes/pseudobulk.ipynb
I have used muscat before and it wraps DESeq2, EdgeR etc for mutli-sample, multi-condition differential analysis. I recommend you to use it.
You can watch the video for this tutorial too: