Introductioon
In scRNA-seq data analysis, one of the most crucial and demanding tasks is determining the optimal resolution and cluster number. Achieving an appropriate balance between over-clustering and under-clustering is often intricate, as it directly impacts the identification of distinct cell populations and biological insights.
The clustering algorithms have many parameters to tune and it can generate more clusters if e.g., you increase the resolution parameter. However, whether the newly generated clusters are meaningful or not is a question.
I previously published a tool scclusteval trying to help with this problem.
Testing scRNAseq clustering significance
Several days ago, I saw this paper came out Significance analysis for clustering with single-cell RNA-sequencing data in Nature Methods. I am eager to give it a try.
The biorxiv version can be found at https://www.biorxiv.org/content/10.1101/2022.08.01.502383v1
The idea is based on the https://github.com/pkimes/sigclust2 which test significant clusters in hierarchical clustering.
However, it assumes an underlying parametric distribution for the data, specifically Gaussian distributions, where distinct populations have different centers. A given set of clusters can then be assessed in a formal and statistically rigorous manner by asking whether or not these clusters could have plausibly arisen under data from a single Gaussian distribution. If so, then the set of clusters likely indicates over-clustering.
Significance of Hierarchical Clustering (SHC) addresses this limitation by incorporating hypothesis testing within the hierarchical procedure. However, SHC is not directly applicable to scRNA-seq data due to the Gaussian distributional assumption, which is inappropriate for these sparse count data.
scSHC implements the significance testing extending to single cell data which is sparse. It models the counts with poisson distribution and use a permutation test to test the significance for the testing statistics: Silhouette width.
To calculate a Silhouette width score for a member of a cluster, you first calculate the average within cluster distances C(i) and the average closest neighbor distance N(i) and use the formula shown in Figure 1 to calculate S(i).
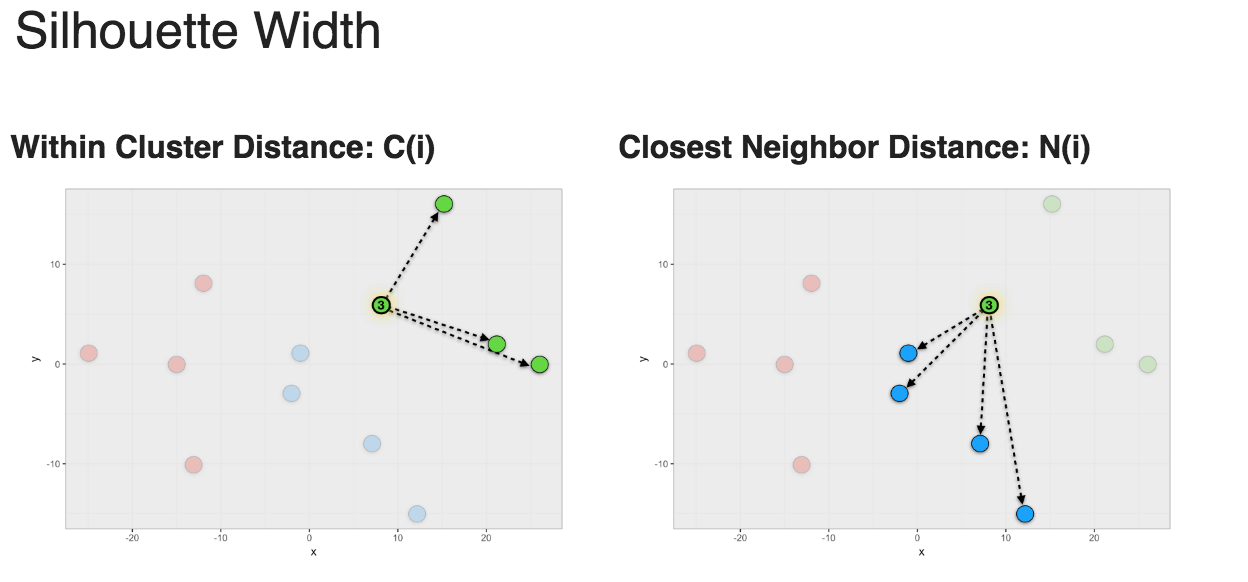
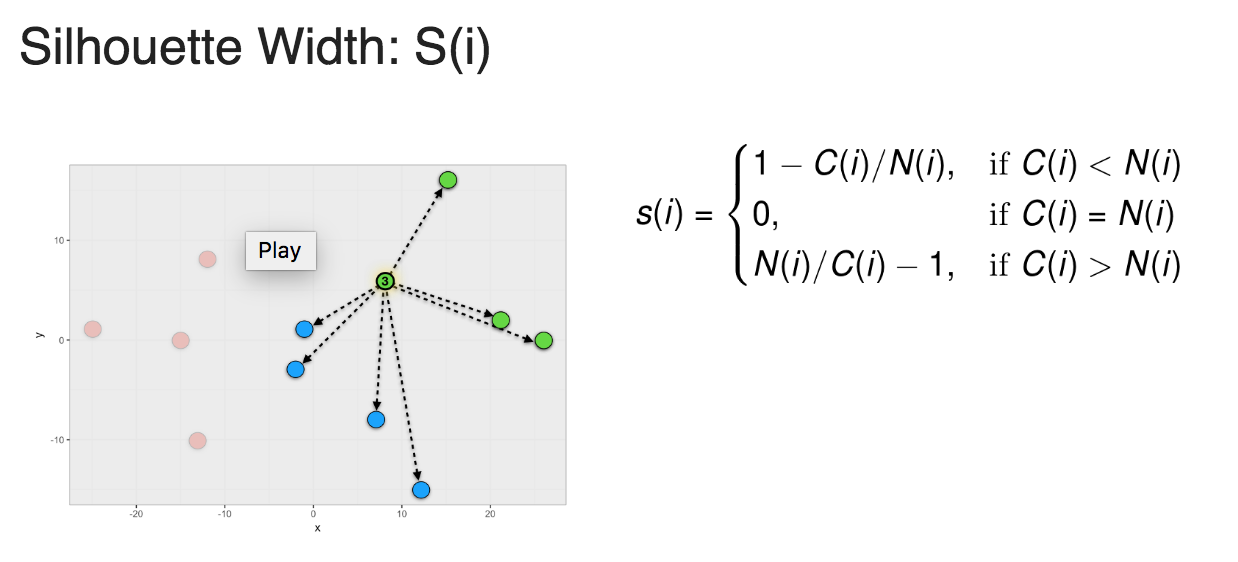
Figure 1: Silhouette width
Silhouette width ranges from -1 to 1. 1 means that member is perfectly match to the cluster it was assigned to. -1 means that member should be placed in the neighboring cluster.
Then, one does hypothesis testing by first stating the null hypothesis:
- H0: there is only one cluster
- H1: there are two clusters
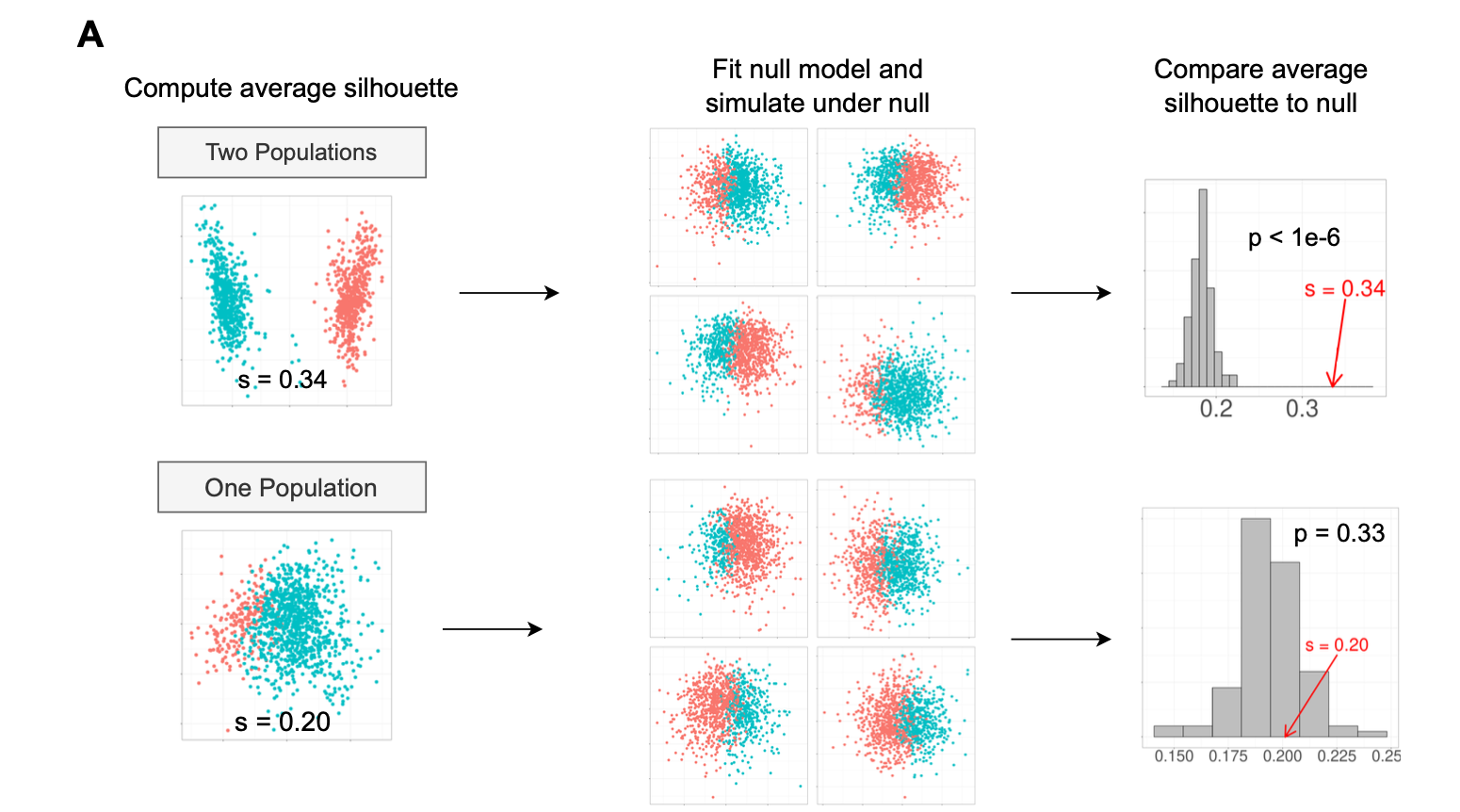
Figure 2: significance test by permutation
The first scenario is that the truth is 2 clusters and we identified two clusters by clustering algorithms. The second scenario is that the truth is only 1 cluster but we also identified two clusters by clustering algorithms.
Under the null hypothesis, one can estimate the distribution parameters and simulate the same distribution by e.g., 100 times and for each time, calculate the Silhouette width for all cells. we plot the simulated average silhouette width (for all cells) as a histogram and then compare the observed silhouette width in the data.
For the two scenarios, we choose a significance level alpha = 0.05.
In the first scenario, the observed silhouette width is greater than all the simulated silhouette width, so we reject the null and accept the alternative: there are two clusters.
In the second scenario, out of 100 permutations, 33 times of the permutation based silhouette score is greater than observed one, so the p-value is 33/100 = 0.33.
It is greater than 0.05, so we fail to reject the null and accept that there is only one cluster in the data.
scSHC tests the clustering significance at every splitting point hierarchically.
install the package
# dependency
BiocManager::install("scry")
devtools::install_github("igrabski/sc-SHC")Load the libraries
library(scSHC)
library(dplyr)
library(Seurat)
library(scCustomize)
library(patchwork)
library(ggplot2)
library(ComplexHeatmap)
library(SeuratData)
set.seed(1234)data("pbmc3k")
pbmc3k#> An object of class Seurat
#> 13714 features across 2700 samples within 1 assay
#> Active assay: RNA (13714 features, 0 variable features)routine processing
pbmc3k<- pbmc3k %>%
NormalizeData(normalization.method = "LogNormalize", scale.factor = 10000) %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
ScaleData() %>%
RunPCA(verbose = FALSE) %>%
FindNeighbors(dims = 1:10, verbose = FALSE) %>%
FindClusters(resolution = 0.5, verbose = FALSE) %>%
RunUMAP(dims = 1:10, verbose = FALSE)
pbmc3k<- pbmc3k[, !is.na(pbmc3k$seurat_annotations)]
p1<- DimPlot_scCustom(pbmc3k, reduction = "umap", label = TRUE, group.by =
"RNA_snn_res.0.5")
p2<- DimPlot_scCustom(pbmc3k, reduction = "umap", label = TRUE, group.by = "seurat_annotations", label.size = 3)
p1 + p2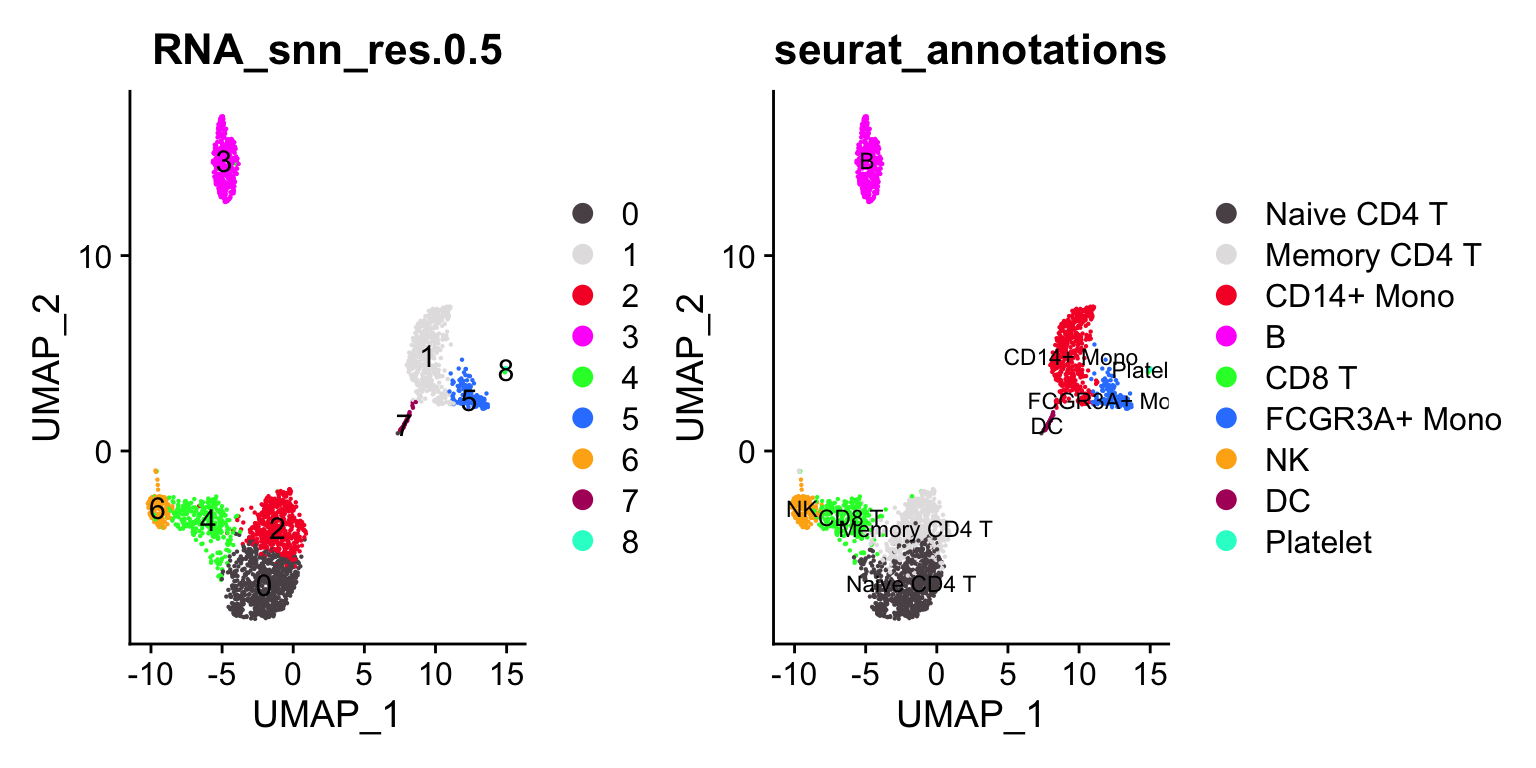
janitor::tabyl(pbmc3k@meta.data, seurat_annotations, RNA_snn_res.0.5)#> seurat_annotations 0 1 2 3 4 5 6 7 8
#> Naive CD4 T 608 0 66 0 23 0 0 0 0
#> Memory CD4 T 69 0 396 0 16 0 2 0 0
#> CD14+ Mono 0 472 0 0 0 3 0 4 1
#> B 0 0 0 343 1 0 0 0 0
#> CD8 T 2 0 3 0 265 0 1 0 0
#> FCGR3A+ Mono 0 7 0 0 0 155 0 0 0
#> NK 0 0 0 0 16 0 139 0 0
#> DC 0 0 0 0 0 0 0 32 0
#> Platelet 0 0 0 0 0 0 0 0 14Let’s artificially increase the resolution (to 1) to over-cluster it.
## artificially increase the resolution
pbmc3k<- pbmc3k %>%
FindNeighbors(dims = 1:10, verbose = FALSE) %>%
FindClusters(resolution = 1, verbose = FALSE)
p3<- DimPlot_scCustom(pbmc3k, reduction = "umap", label = TRUE, group.by = "RNA_snn_res.1")
(p1 + p3) / p2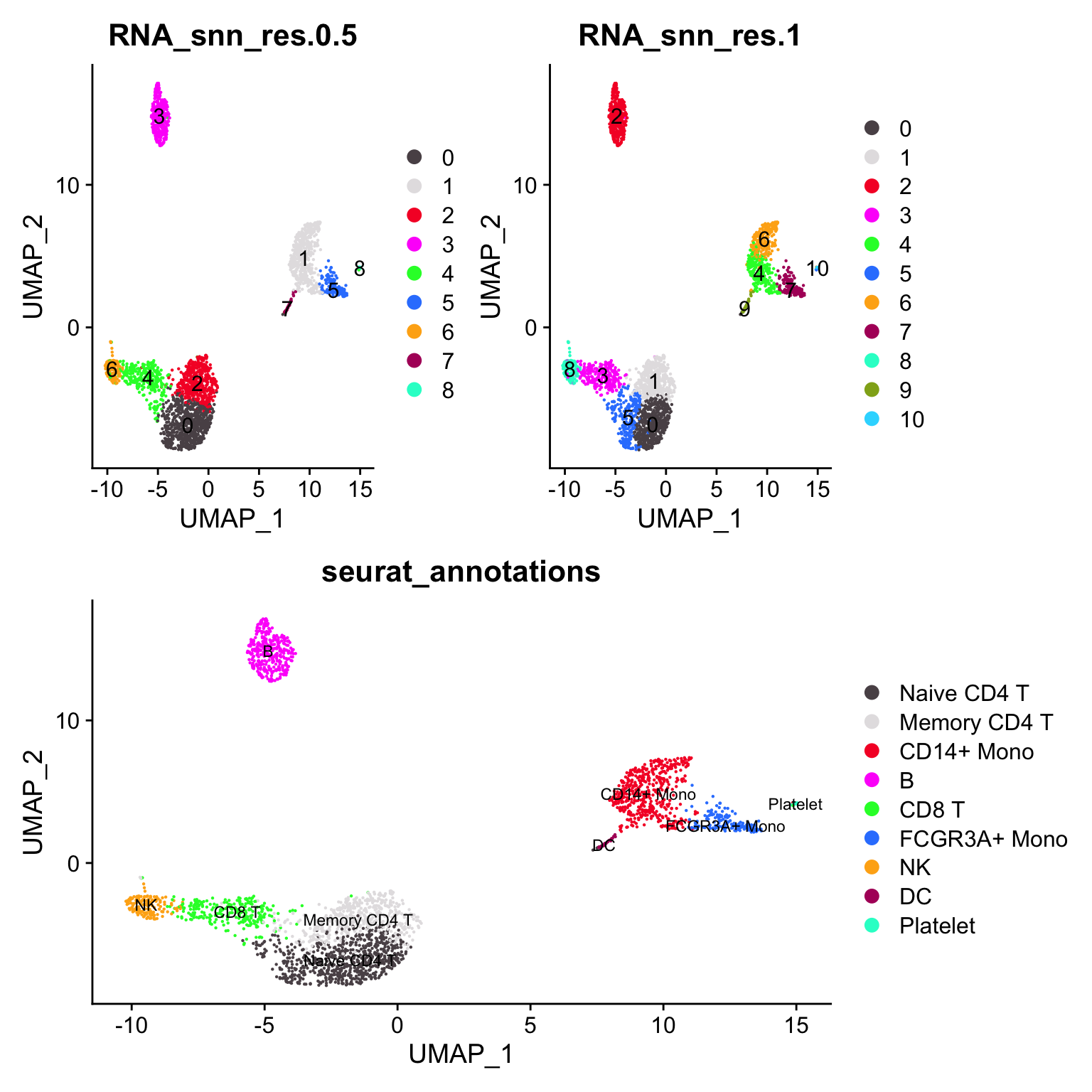
CD4 naive cell cluster is split to 2 clusters (0 -> 0, 5) the CD14+ monocyte cluster is split into 2 clusters (1 -> 4, 6)
janitor::tabyl(pbmc3k@meta.data, RNA_snn_res.1, RNA_snn_res.0.5)#> RNA_snn_res.1 0 1 2 3 4 5 6 7 8
#> 0 470 0 49 0 0 0 0 0 0
#> 1 3 0 413 0 1 0 0 0 0
#> 2 0 0 0 343 0 0 0 0 0
#> 3 0 0 0 0 279 0 0 0 0
#> 4 0 262 0 0 0 1 0 0 0
#> 5 206 0 3 0 41 0 0 0 0
#> 6 0 217 0 0 0 0 0 0 1
#> 7 0 0 0 0 0 157 0 0 0
#> 8 0 0 0 0 0 0 142 0 0
#> 9 0 0 0 0 0 0 0 36 0
#> 10 0 0 0 0 0 0 0 0 14use scSHC to do the clustering
library(tictoc)
tic()
clusters<- scSHC(pbmc3k@assays$RNA@counts, cores = 6,
num_PCs = 30,
num_features = 2000)
toc()#> 162.082 sec elapsedvisualize the data
The scSHC output is a list of 2 elements, the first element contains the cluster id for each cell and the second element contains
table(clusters[[1]])#>
#> 1 2 3 4 5 6 7 8
#> 448 105 130 154 521 334 426 520# the second
clusters[[2]]#> levelName
#> 1 Node 0: 0
#> 2 ¦--Node 1: 0
#> 3 ¦ ¦--Node 3: 0
#> 4 ¦ ¦ ¦--Cluster 2: 1
#> 5 ¦ ¦ °--Cluster 3: 1
#> 6 ¦ °--Cluster 1: 1
#> 7 °--Node 2: 0
#> 8 ¦--Node 4: 0
#> 9 ¦ ¦--Cluster 4: 1
#> 10 ¦ °--Cluster 5: 1
#> 11 °--Node 5: 0
#> 12 ¦--Cluster 6: 1
#> 13 °--Node 6: 0.01
#> 14 ¦--Cluster 7: 1
#> 15 °--Cluster 8: 1clusters[[1]] %>%
tibble::enframe() %>%
head()#> # A tibble: 6 × 2
#> name value
#> <chr> <dbl>
#> 1 AAACATACAACCAC 5
#> 2 AAACATTGAGCTAC 6
#> 3 AAACATTGATCAGC 5
#> 4 AAACCGTGCTTCCG 3
#> 5 AAACCGTGTATGCG 4
#> 6 AAACGCACTGGTAC 5scSHC_clusters<- clusters[[1]] %>%
tibble::enframe(name = "barcode", value = "scSHC_clusters")
scSHC_clusters<- as.data.frame(scSHC_clusters)
rownames(scSHC_clusters)<- scSHC_clusters$barcode
head(scSHC_clusters)#> barcode scSHC_clusters
#> AAACATACAACCAC AAACATACAACCAC 5
#> AAACATTGAGCTAC AAACATTGAGCTAC 6
#> AAACATTGATCAGC AAACATTGATCAGC 5
#> AAACCGTGCTTCCG AAACCGTGCTTCCG 3
#> AAACCGTGTATGCG AAACCGTGTATGCG 4
#> AAACGCACTGGTAC AAACGCACTGGTAC 5add the scSHC cluster to the seurat metadata slot
pbmc3k<- AddMetaData(pbmc3k, metadata = scSHC_clusters)
pbmc3k@meta.data %>% head()#> orig.ident nCount_RNA nFeature_RNA seurat_annotations
#> AAACATACAACCAC pbmc3k 2419 779 Memory CD4 T
#> AAACATTGAGCTAC pbmc3k 4903 1352 B
#> AAACATTGATCAGC pbmc3k 3147 1129 Memory CD4 T
#> AAACCGTGCTTCCG pbmc3k 2639 960 CD14+ Mono
#> AAACCGTGTATGCG pbmc3k 980 521 NK
#> AAACGCACTGGTAC pbmc3k 2163 781 Memory CD4 T
#> RNA_snn_res.0.5 seurat_clusters RNA_snn_res.1 barcode
#> AAACATACAACCAC 0 5 5 AAACATACAACCAC
#> AAACATTGAGCTAC 3 2 2 AAACATTGAGCTAC
#> AAACATTGATCAGC 2 1 1 AAACATTGATCAGC
#> AAACCGTGCTTCCG 5 7 7 AAACCGTGCTTCCG
#> AAACCGTGTATGCG 6 8 8 AAACCGTGTATGCG
#> AAACGCACTGGTAC 2 1 1 AAACGCACTGGTAC
#> scSHC_clusters
#> AAACATACAACCAC 5
#> AAACATTGAGCTAC 6
#> AAACATTGATCAGC 5
#> AAACCGTGCTTCCG 3
#> AAACCGTGTATGCG 4
#> AAACGCACTGGTAC 5all.equal(rownames(pbmc3k@meta.data), unname(pbmc3k$barcode))#> [1] TRUEplotting
p4<- DimPlot_scCustom(pbmc3k, group.by = "scSHC_clusters")
(p2 + p4) / (p1 + p3)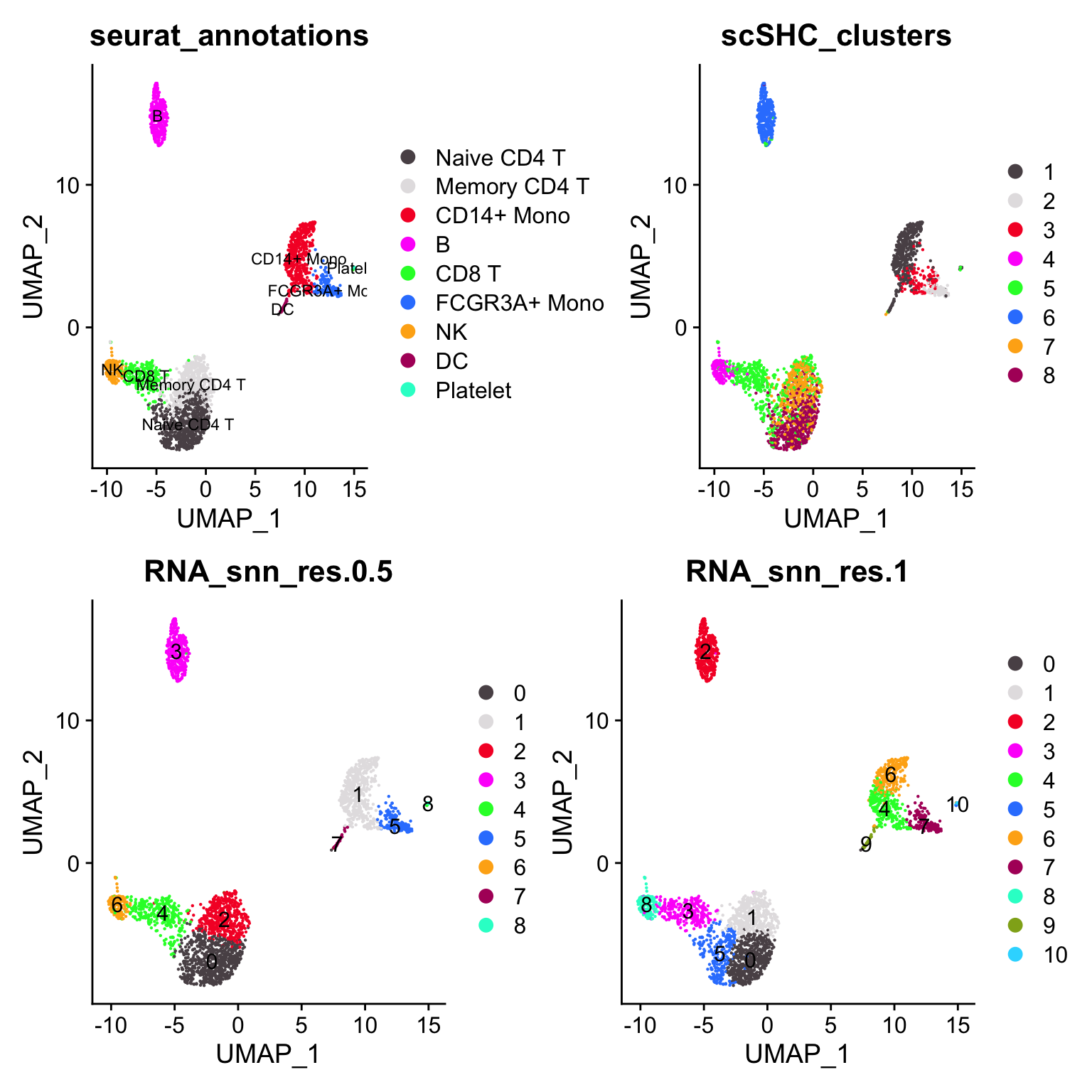
janitor::tabyl(pbmc3k@meta.data, seurat_annotations, scSHC_clusters)#> seurat_annotations 1 2 3 4 5 6 7 8
#> Naive CD4 T 0 0 0 0 80 0 150 467
#> Memory CD4 T 0 0 0 0 161 0 274 48
#> CD14+ Mono 398 1 81 0 0 0 0 0
#> B 0 0 0 0 8 334 1 1
#> CD8 T 0 0 0 11 255 0 1 4
#> FCGR3A+ Mono 9 104 49 0 0 0 0 0
#> NK 0 0 0 143 12 0 0 0
#> DC 29 0 0 0 3 0 0 0
#> Platelet 12 0 0 0 2 0 0 0What surprises me is that scSHC does not separate the CD14+ monocytes and the dendritic cells. (cluster 1 is a mix of CD14+ monocytes, DCs and platelet)
conclusions
One may fine tune
num_featuresandnum_PCsto get a better results. My conclusion is that although it seems statistically attractive, significance testing for scRNAseq clustering is still an unsolved problem. Cell type or cell state is inherently complicated and sometimes in a continuum.Also, we do not have a ground truth here. The Seurat cell annotations provided by the developers may not be 100% correct.
It took ~3mins using 6 CPUs for 3000 cells. It can take long time if you have a lot of cells and many clusters as
scSHCtest the cluster significance at every splitting node.
Further reading
what is a cell type? https://twitter.com/tangming2005/status/1680932947619201025
A reference cell tree will serve science better than a reference cell atlas
The evolving concept of cell identity in the single cell era
Bonus, a blog post by Matthew Bernstein On cell types and cell states https://mbernste.github.io/posts/cell_types_cell_states/