Following my last blog post, download the CITE-seq protein and RNA count data at here.
library(Seurat)
library(ggplot2)
library(dplyr)
pbmc<- readRDS("~/blog_data/CITEseq/pbmc1k_adt.rds")How to normalize protein ADT count data?
Seurat uses the centered log ratio (CLR) to normalize protein count data.
In the Seurat github page:
# https://github.com/satijalab/seurat/blob/fc4a4f5203227832477a576bfe01bc6efeb23f51/R/preprocessing.R#L1768-L1769
clr_function <- function(x) {
return(log1p(x = x / (exp(x = sum(log1p(x = x[x > 0]), na.rm = TRUE) / length(x = x)))))
}log1p(x) computes log(1+x) accurately also for |x| << 1.
It addes a pseduo-count in the last step by using log1p and it effectively makes the transformed values >=0.
Let’s see it in action.
pbmc <- NormalizeData(pbmc, normalization.method = "CLR", margin = 2, assay = "ADT")
# Now, we will visualize CD14 levels for RNA and protein By setting the default assay, we can
# visualize one or the other
DefaultAssay(pbmc) <- "ADT"
FeaturePlot(pbmc, "CD19", cols = c("lightgrey", "darkgreen")) + ggtitle("CD19 protein")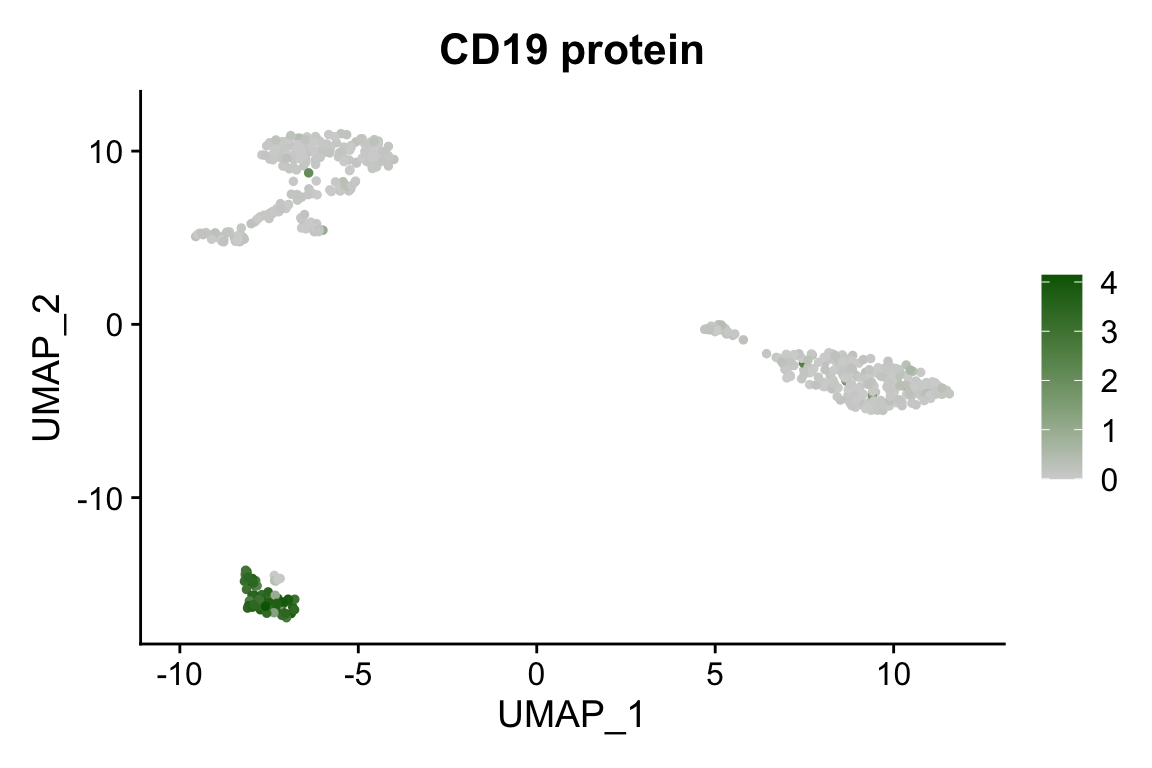
RidgePlot(pbmc, features = c("CD19", "CD3"))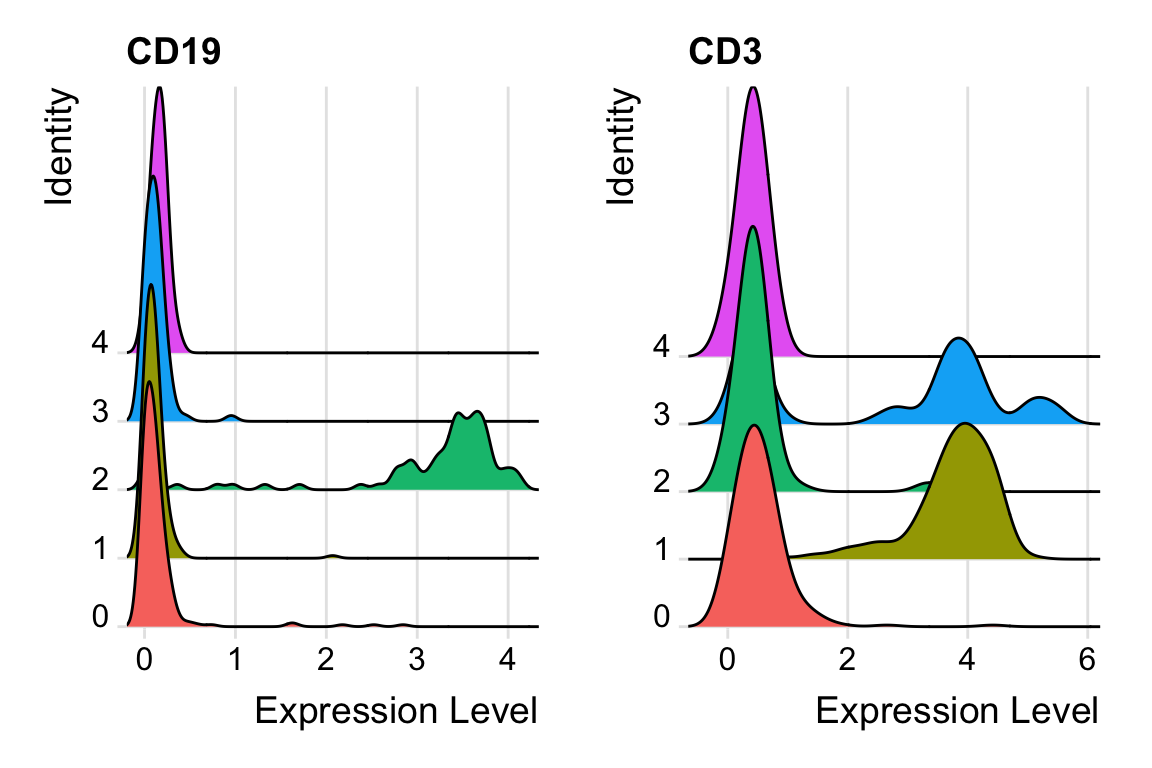 We do see the minimal value is 0.
We do see the minimal value is 0.
Second, what doesmargin = 2 mean?
- margin=1 means “perform CLR normalization for each feature independently”
- margin=2 means “perform CLR normalization within a cell”
Let’s verify it.
# raw counts
adt_count<- pbmc@assays$ADT@counts
# first cell
adt_count[,1]#> CD3 CD4 CD8a CD14 CD15 CD16 CD56 CD19 CD25 CD45RA CD45RO
#> 4 483 16 1148 36 12 18 1 4 248 290
#> PD-1 TIGIT CD127 IgG2a IgG1 IgG2b
#> 12 4 5 4 6 2#normalized data
adt_norm<- pbmc@assays$ADT@data
# first cell
adt_norm[,1]#> CD3 CD4 CD8a CD14 CD15 CD16 CD56
#> 0.18254021 3.22611701 0.58836964 4.06860466 1.03046254 0.47049554 0.64247520
#> CD19 CD25 CD45RA CD45RO PD-1 TIGIT CD127
#> 0.04885264 0.18254021 2.59646813 2.74206669 0.47049554 0.18254021 0.22340593
#> IgG2a IgG1 IgG2b
#> 0.18254021 0.26266700 0.09542945all.equal(clr_function(adt_count[,1]), adt_norm[,1])#> [1] TRUEThe result shows that when specifying “margin = 2”, seurat is normalizing all the features within a cell.
However, the help page of the normalization is a little bit confusing.
margin If performing CLR normalization, normalize across features (1) or cells (2)
and when you invoke the function, if you specify marigin =1 it will message: “Normalizing across features”; if you specify margin = 2, it will message “Normalizing across cells”.
This is oppostite to what seurat is doing internally. Read this issue and someone else had the same intepretation https://github.com/satijalab/seurat/issues/3605.
CLR normalization in detail
Now, let’s take a closer look at centered log-ratio (CLR) normalization. CLR was initially used in this paper Simultaneous epitope and transcriptome measurement in single cells on compositional data.

The first function below is what the orginal paper uses.
## the usual CLR adding pseduo-count for each protein first
clr_original <- function(x) {
return(log((x + 1) / mean(x + 1, na.rm = TRUE)))
}
## clr from seurat
clr_function <- function(x) {
return(log1p(x = x / (exp(x = sum(log1p(x = x[x > 0]), na.rm = TRUE) / length(x = x)))))
}Seurat CLR removes 0 counts first by x[x>0] and then log1p transform the raw counts, sum them up, calculate the average of the log counts, exp it back, and then divided the raw counts by this average and finally log1p again…
Seurat CLR normalization.
clr_norm<- apply(adt_count, MARGIN = 2, FUN= clr_function)
# indeed, it is the same with the normalized ADT data in the seurat object
all.equal(clr_norm, pbmc@assays$ADT@data)#> [1] TRUErange(clr_norm)#> [1] 0.000000 5.754108hist(clr_norm)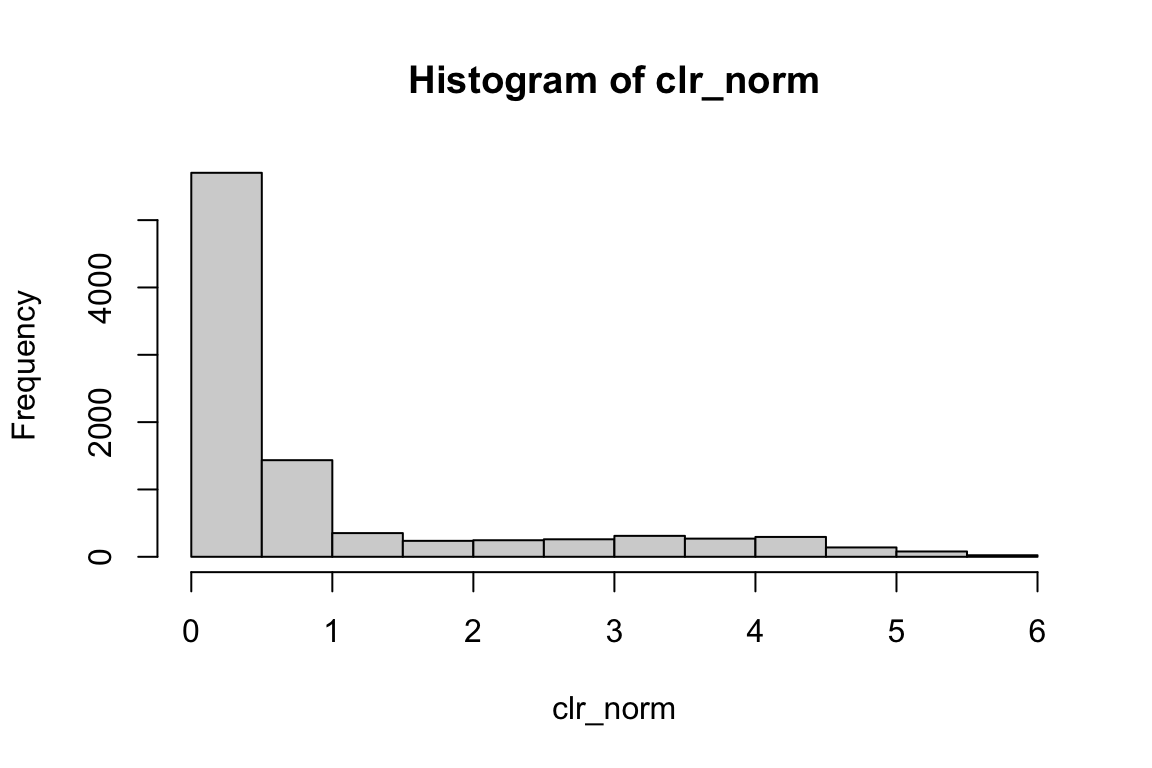 We do not see negative values.
We do not see negative values.
The original CLR
clr_original_norm<- apply(adt_count, MARGIN = 2, clr_original)
range(clr_original_norm)#> [1] -6.363028 2.786000hist(clr_original_norm)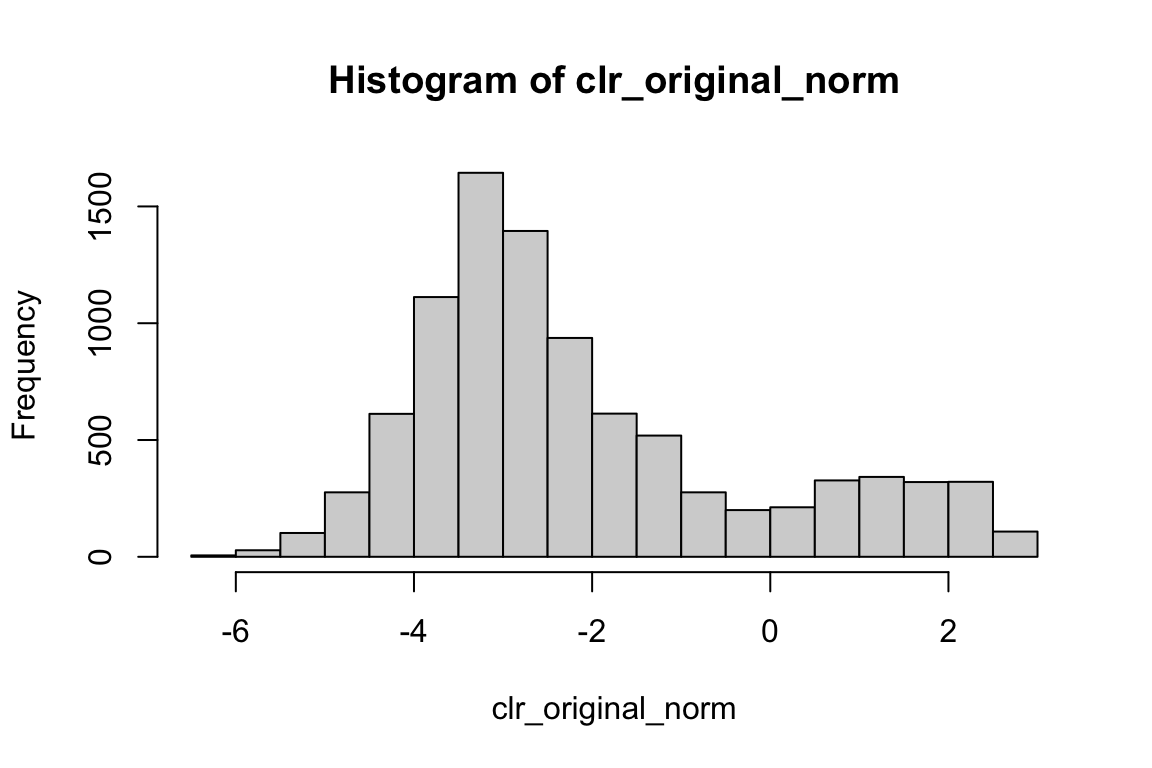 We do see negative values.
We do see negative values.
Visualize in Ridge plots:
get_expression_meta<- function(obj, mat){
meta<- obj@meta.data %>%
tibble::rownames_to_column(var = "cell")
df<- t(mat) %>%
as.data.frame() %>%
tibble::rownames_to_column(var = "cell") %>%
left_join(meta)
return(df)
}
p1<- get_expression_meta(pbmc, clr_norm ) %>%
ggplot(aes(x = CD19, y = celltypes)) +
ggridges::geom_density_ridges(aes(fill = celltypes)) +
theme_bw(base_size = 14) +
xlab("") +
ylab("") +
ggtitle("Seurat CLR")
p2<- get_expression_meta(pbmc, clr_original_norm ) %>%
ggplot(aes(x = CD19, y = celltypes)) +
ggridges::geom_density_ridges(aes(fill = celltypes)) +
theme_bw(base_size = 14) +
xlab("") +
ylab("") +
ggtitle("original CLR")
p1 + p2 + patchwork::plot_layout(guides = "collect")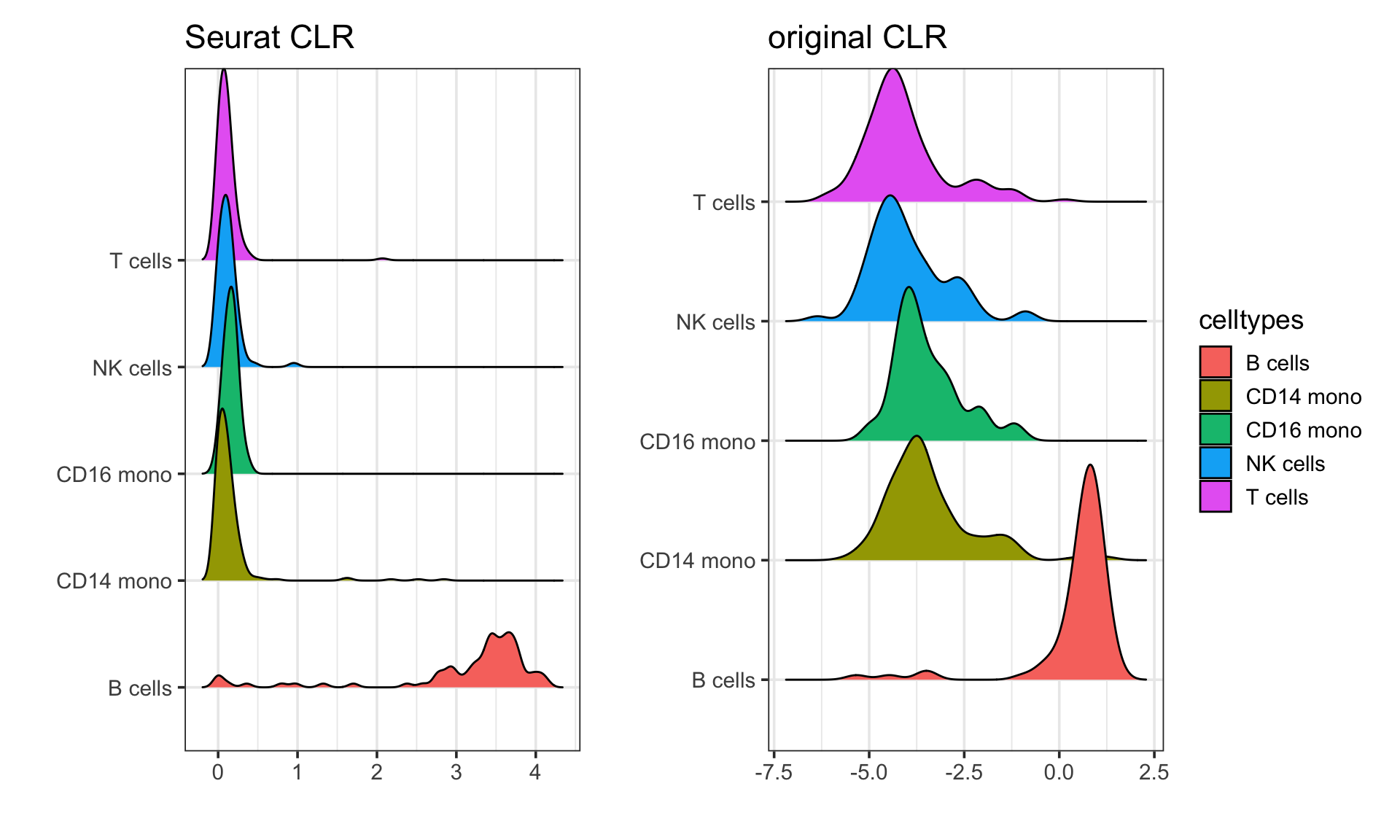
visualize in Heatmap
library(ComplexHeatmap)
hp1<- Heatmap(clr_norm,
show_column_names = FALSE,
name = "Seurat CLR")
row_index<- row_order(hp1)
col_index<- column_order(hp1)
hp2<- Heatmap(clr_original_norm,
show_column_names = FALSE,
row_order = row_index,
column_order = col_index,
name = "original CLR")
hp1 + hp2 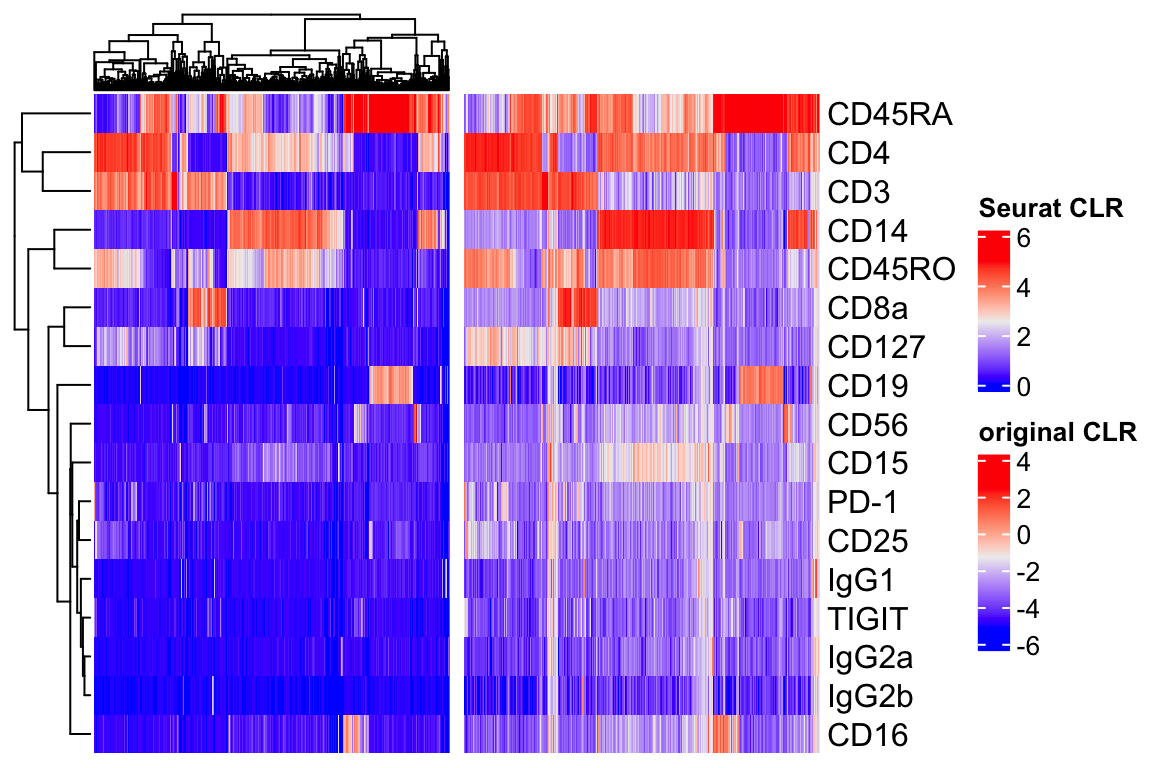
They both look similar in terms of patterns. The Seurat CLR makes it 0 bound and the authors argue it is better for visualization.
Take home messages
Details matter. If you blindly uses the functions in a package, you may get the wrong interpretation of your results.
Documentation matters. If the documentation is confusing, it may lead to unintended usages of the options.