Introduction
A state-of-the-art method called CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing) allows surface protein levels and RNA expression to be measured simultaneously in individual cells. CITE-seq uses traditional single-cell RNA-sequencing to read out both transcriptome and proteomic information from the same cell after labeling it with oligo-conjugated antibodies. This gets over the drawbacks of techniques that just test proteins or RNA separately. CITE-seq reveals coordinated control of gene and protein activity, offering a potent multidimensional perspective of cell states. Read more from https://cite-seq.com/
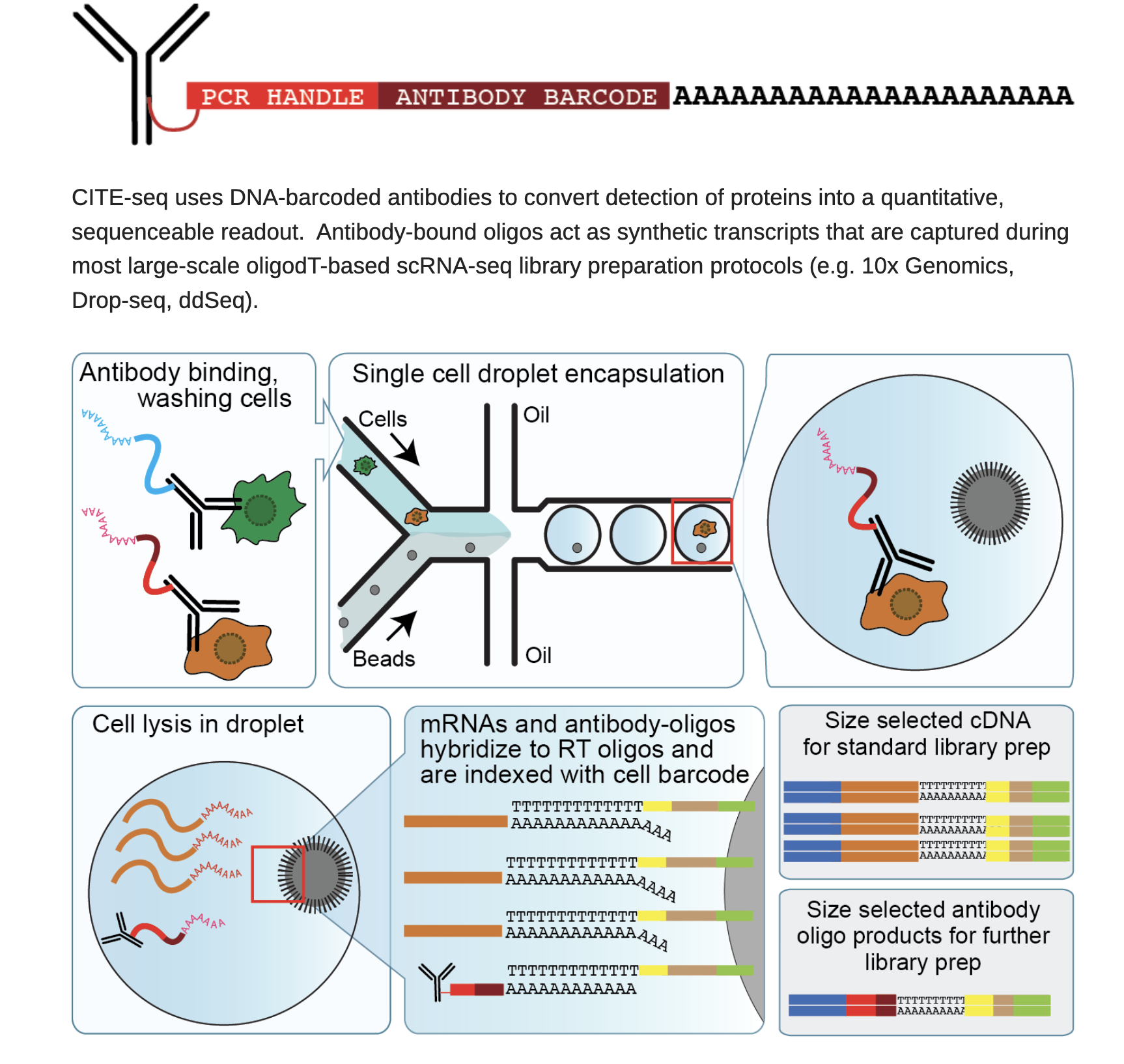
Given that protein and RNA levels are not always correlated. It is important to get a smaller panel of protein expression profile. In addition, most of the scRNAseq can not differentiate isoforms such as CD45RA and CD45RO which marks different state of T cells.
CD45 (official gene name PTPRC) is an abundant leukocyte surface marker that exists in several isoforms generated by alternative splicing of exons 4, 5, and 6. The two main isoforms are:
CD45RA - Contains exon 4 but lacks exons 5 and 6. It is expressed on naive and some memory T cells, naive B cells. CD45RA is important for maintaining a naive phenotype. CD45RA+ cells tend to be naive cells that have not encountered antigen before. They require stronger activation signals to become activated.
CD45RO - Lacks exon 4 but contains exons 5 and 6. It is expressed on most memory T cells, memory B cells, macrophages, and activated NK cells. CD45RO is associated with antigen-experienced cells. CD45RO+ cells are antigen-experienced cells that can respond faster and be activated by lower activation thresholds. They play a major role in secondary immune responses. So in essence:
In this blog post, I will walk you through the pre-processing steps from fastq files to the count matrix for the RNA and the protein (antibody derived tag/ADT count) using Alevin which is part of the Salmon.
Other solutions:
Tutorial references
The authors of Alevin created Simpleaf, which is a command line toolkit written in rust that exposes a unified and simplified interface for processing scRNA-seq datasets using the alevin-fry ecosystem of tools.
- This is the most recent tutorial using
simpleafhttps://combine-lab.github.io/alevin-fry-tutorials/2023/running-simpleaf-workflow/. It involves making a configuration of a json like file. I prefer to still use the commands instead, so I read
some old tutorials:
- https://combine-lab.github.io/alevin-fry-tutorials/2021/af-feature-bc/
- https://combine-lab.github.io/alevin-tutorial/2020/alevin-features/
and put the command together below.
Installation and setup
Install simpleaf by conda.
# on my local computer
gcloud compute ssh mtang@machine-name
# on google VM, attach disk to the compute VM
sudo mount -o discard,defaults /dev/sdb /mnt/disks/tommy
# install via conda
conda create -n af -y -c bioconda -c conda-forge simpleaf
conda activate afset up the variables
cd /mnt/disks/tommy/
export AF_SAMPLE_DIR=$PWD/af_test_workdir
mkdir $AF_SAMPLE_DIR
FASTQ_DIR="$AF_SAMPLE_DIR/data/pbmc_1k_protein_v3_fastqs"
mkdir -p $FASTQ_DIRdefine env variable
export ALEVIN_FRY_HOME="$AF_SAMPLE_DIR/af_home"
simpleaf configuration
simpleaf set-pathsDownload RNA reference genome and gene annotations
cd $AF_SAMPLE_DIR
REF_DIR="$AF_SAMPLE_DIR/data/refdata-gex-GRCh38-2020-A"
mkdir -p $REF_DIR
IDX_DIR="$AF_SAMPLE_DIR/human-2020-A_splici"
# Download reference genome and gene annotations
wget -qO- https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2020-A.tar.gz | tar xzf - --strip-components=1 -C $REF_DIRBuild gene expression index
## make sure the read length rlen is set correctly, I went to the fastq and wc -L the first read
simpleaf index \
--output $IDX_DIR \
--fasta $REF_DIR/fasta/genome.fa \
--gtf $REF_DIR/genes/genes.gtf \
--rlen 90 \
--threads 16 Download feature reference and build the index for the protein
# feature barcode reference
FEATURE_REF_PATH="$AF_SAMPLE_DIR/data/feature_reference.csv"
wget -v -O $FEATURE_REF_PATH -L \
https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_protein_v3/pbmc_1k_protein_v3_feature_ref.csv
head feature_reference.csv
id,name,read,pattern,sequence,feature_type
CD3,CD3_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,AACAAGACCCTTGAG,Antibody Capture
CD4,CD4_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,TACCCGTAATAGCGT,Antibody Capture
CD8a,CD8a_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,ATTGGCACTCAGATG,Antibody Capture
CD14,CD14_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,GAAAGTCAAAGCACT,Antibody Capture
CD15,CD15_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,ACGAATCAATCTGTG,Antibody Capture
CD16,CD16_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,GTCTTTGTCAGTGCA,Antibody Capture
CD56,CD56_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,GTTGTCCGACAATAC,Antibody Capture
CD19,CD19_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,TCAACGCTTGGCTAG,Antibody Capture
CD25,CD25_TotalSeqB,R2,5PNNNNNNNNNN(BC)NNNNNNNNN,GTGCATTCAACAGTA,Antibody Capture
#only need the first and the fifth column
cat feature_reference.csv| awk -F "," '{print $1"\t"$5}’ | tail -n +2 > adt.tsv
# build index for the adt
salmon index -t adt.tsv -i adt_index --features -k7
## gene to transcript mapping, it is the same column 1
awk '{print $1"\t"$1;}' adt.tsv > t2g_adt.tsvDownload FASTQ files
We will use the 1k PBMC dataset from 10x
https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_protein_v3/pbmc_1k_protein_v3_fastqs.tar
After unzipping, it has two folders of fastqs, one for RNA, one for protein ADT.
wget -qO- https://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_1k_protein_v3/pbmc_1k_protein_v3_fastqs.tar | \
tar xf - --strip-components=1 -C $FASTQ_DIR
# take a look at the files
ls pbmc_1k_protein_v3_fastqs/*
pbmc_1k_protein_v3_fastqs/pbmc_1k_protein_v3_antibody_fastqs:
pbmc_1k_protein_v3_antibody_S2_L001_I1_001.fastq.gz pbmc_1k_protein_v3_antibody_S2_L002_I1_001.fastq.gz
pbmc_1k_protein_v3_antibody_S2_L001_R1_001.fastq.gz pbmc_1k_protein_v3_antibody_S2_L002_R1_001.fastq.gz
pbmc_1k_protein_v3_antibody_S2_L001_R2_001.fastq.gz pbmc_1k_protein_v3_antibody_S2_L002_R2_001.fastq.gz
pbmc_1k_protein_v3_fastqs/pbmc_1k_protein_v3_gex_fastqs:
pbmc_1k_protein_v3_gex_S1_L001_I1_001.fastq.gz pbmc_1k_protein_v3_gex_S1_L001_R2_001.fastq.gz pbmc_1k_protein_v3_gex_S1_L002_R1_001.fastq.gz
pbmc_1k_protein_v3_gex_S1_L001_R1_001.fastq.gz pbmc_1k_protein_v3_gex_S1_L002_I1_001.fastq.gz pbmc_1k_protein_v3_gex_S1_L002_R2_001.fastq.gzQuantification
quantify RNA
# this unix command trick is to find the fastq file path and stitch them by ,
reads1_pat="_R1_"
reads1="$(find -L ${FASTQ_DIR}/pbmc_1k_protein_v3_gex_fastqs -name "*$reads1_pat*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd, -)"
reads2_pat="_R2_"
reads2="$(find -L ${FASTQ_DIR}/pbmc_1k_protein_v3_gex_fastqs -name "*$reads2_pat*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd, -)"
simpleaf quant \
--reads1 $reads1 \
--reads2 $reads2 \
--threads 16 \
--index $IDX_DIR/index \
--chemistry 10xv3 --resolution cr-like \
--expected-ori fw --unfiltered-pl \
--t2g-map $IDX_DIR/index/t2g_3col.tsv \
--output alevin_rna
The transcript to gene mapping file t2g_3col.tsv was generated when making the index file by
simpleaf index.
Note, for 10xv2 5’ data, change the argument to --expected-ori cr.
see https://github.com/COMBINE-lab/alevin-fry/issues/118 and
quantify protein adt
reads1_pat="_R1_"
reads1="$(find -L ${FASTQ_DIR}/pbmc_1k_protein_v3_antibody_fastqs -name "*$reads1_pat*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd, -)"
reads2_pat="_R2_"
reads2="$(find -L ${FASTQ_DIR}/pbmc_1k_protein_v3_antibody_fastqs -name "*$reads2_pat*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd, -)"
simpleaf quant \
--reads1 $reads1 \
--reads2 $reads2 \
--threads 16 \
--index $AF_SAMPLE_DIR/data/adt_index \
--chemistry 10xv3 --resolution cr-like \
--expected-ori fw --unfiltered-pl \
--t2g-map $AF_SAMPLE_DIR/data/t2g_adt.tsv \
--output alevin_adtSpecify --chemistry 10xv3 somehow just works. Just to be clear, if you know where
the feature barcode sequences are in the reads, specify e.g., --chemistry 1{b[16]u[10]x:}2{x[10]r[15]x:} exactly and provide the path for the whitelist:
--unfiltered-pl /mnt/disks/tommy/af_test_workdir/737K-august-2016.txt.
see https://github.com/COMBINE-lab/alevin-fry/issues/136
Hoary! Now we have the count matrix for the RNA and the protein ready for downstream analysis. Stay tuned for my next blog post!