 My caption 😄
My caption 😄
Fast analysis of scATAC-seq data using a predefined set of genomic regions
Abstract
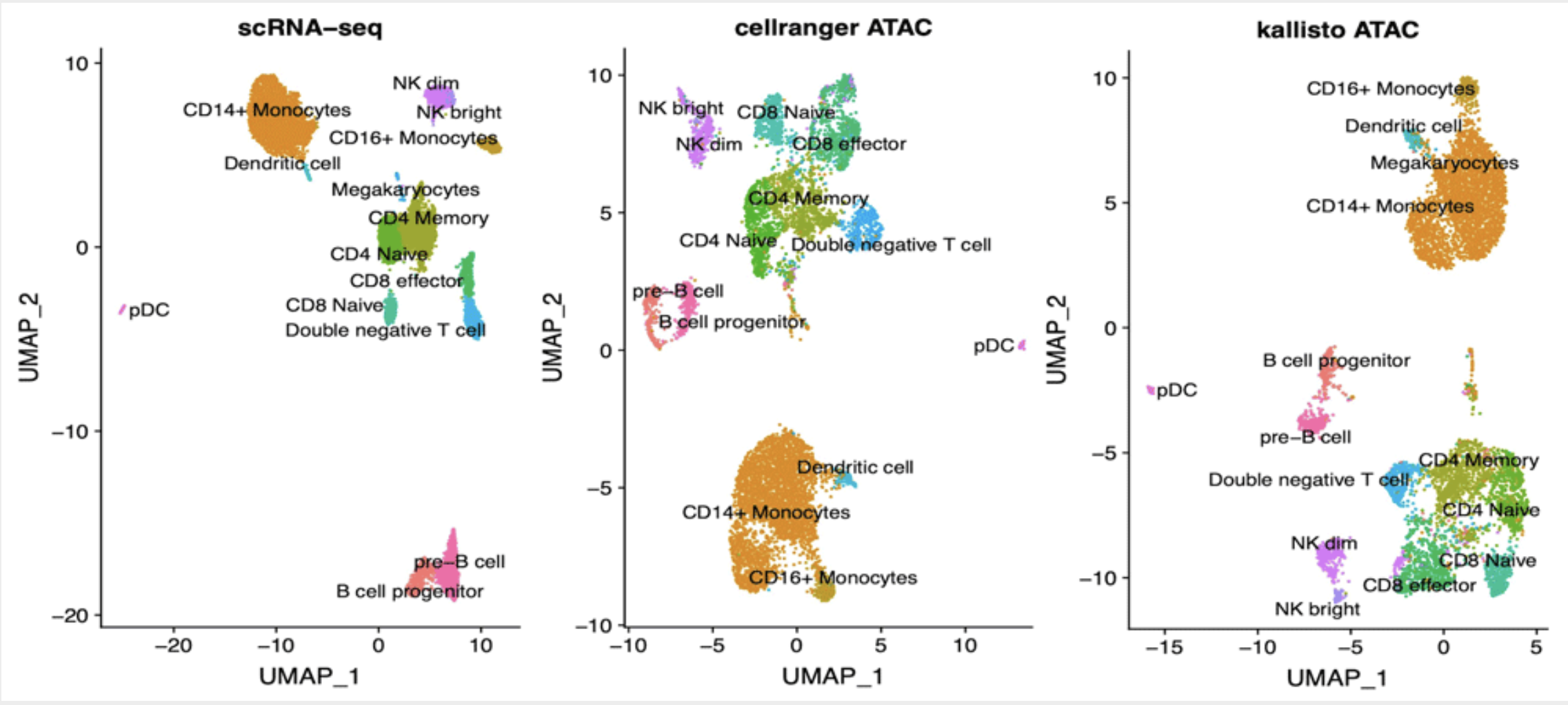
Background: Analysis of scATAC-seq data has been recently scaled to thousands of cells. While processing of other types of single cell data was boosted by the implementation of alignment-free techniques, pipelines available to process scATAC-seq data still require large computational resources. We propose here an approach based on pseudoalignment, which reduces the execution times and hardware needs at little cost for precision. Methods: Public data for 10k PBMC were downloaded from 10x Genomics web site. Reads were aligned to various references derived from DNase I Hypersensitive Sites (DHS) using kallisto and quantified with bustools. We compared our results with the ones publicly available derived by cellranger-atac. Results: We found that kallisto does not introduce biases in quantification of known peaks and cells groups are identified in a consistent way. We also found that cell identification is robust when analysis is performed using DHS-derived reference in place of de novo identification of ATAC peaks. Lastly, we found that our approach is suitable for reliable quantification of gene activity based on scATAC-seq signal, thus allows for efficient labelling of cell groups based on marker genes.Conclusions: Analysis of scATAC-seq data by means of kallisto produces results in line with standard pipelines while being considerably faster; using a set of known DHS sites as reference does not affect the ability to characterize the cell populations
More detail can easily be written here using Markdown and $\rm \LaTeX$ math code.