Publications
Filter by type:
Targeting the chromatin modifying enzyme, KDM5C, enhances AKT inhibition response in ER+ breast cancer
Capivasertib is an AKT inhibitor approved for the treatment of HR+/HER2- advanced breast cancer with PIK3CA, PTEN, or AKT-1 alterations. To identify resistance mechanisms and combination opportunities, we performed CRISPR screens that revealed loss of chromatin regulators including KDM5C and KAT6A increased sensitivity to capivasertib. KDM5C inhibition increased cell stress, DNA damage, and apoptosis when combined with the AKT inhibitor, with the strongest effects in cancers harboring PIK3CA, PTEN, or AKT-1 alterations. These findings nominate KDM5C as a combination target to enhance AKT inhibition response in ER+ breast cancer.
In Molecular Cancer Therapeutics,
2026
Computational Analyses and Challenges of Single-cell ATAC-seq
Single-cell ATAC-seq is a powerful approach for examining cellular epigenetic variation and gene regulation mechanisms. However, low genomic coverage per cell creates inherent data sparsity and missing-value challenges that require specialized computational solutions. This review synthesizes published analytical workflows spanning preprocessing through downstream analysis, including quality control, alignment, peak identification, dimensionality reduction, clustering, regulatory scoring, cell classification, and data integration. The authors additionally survey key databases, emerging deep-learning techniques, AI foundation models designed for this modality, and recent spatial ATAC-seq advances with corresponding computational tools.
In Genomics, Proteomics & Bioinformatics,
2025
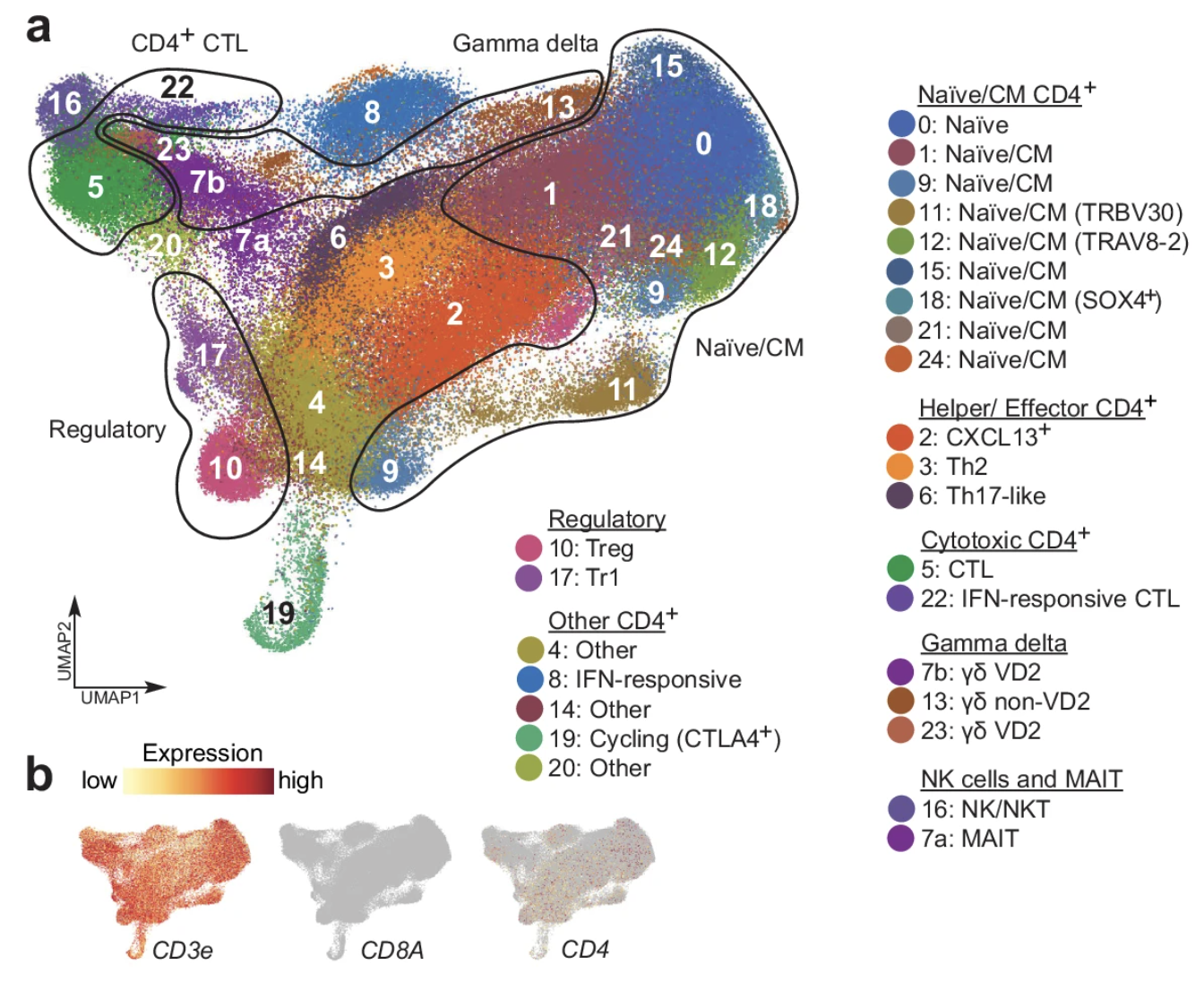
Cancer-specific innate and adaptive immune rewiring drives resistance to PD-1 blockade in classic Hodgkin lymphoma
Hodgkin Reed-Sternberg (HRS) cells of classic Hodgkin lymphoma (cHL), like many solid tumors, elicit ineffective immune responses. However, patients with cHL are highly responsive to PD-1 blockade, which largely depends on HRS cell-specific retention of MHC class II and implicates CD4+ T cells and additional MHC class I-independent immune effectors. Here, we utilize single-cell RNA sequencing and spatial analysis to define shared circulating and microenvironmental features of the immune response to PD-1 blockade in cHL. Compared with non-responders, responding patients have more circulating CD4+ naïve and central memory T cells and B cells, as well as more diverse CD4+ T cell and B cell receptor repertoires. Importantly, a population of circulating and tumor-infiltrating IL1β+ monocytes/macrophages is detectable in patients with cHL but not healthy donors, and a proinflammatory, tumor-promoting signature of these circulating IL1β+ monocytes is associated with resistance to PD-1 blockade in cHL. Altogether, our findings reveal extensive immune rewiring and complementary roles of CD4+ T cells, B cells and IL1β+ monocytes in the response to PD-1 blockade and suggest that these features can be captured with a peripheral blood test.
In Nature Communications,
2024
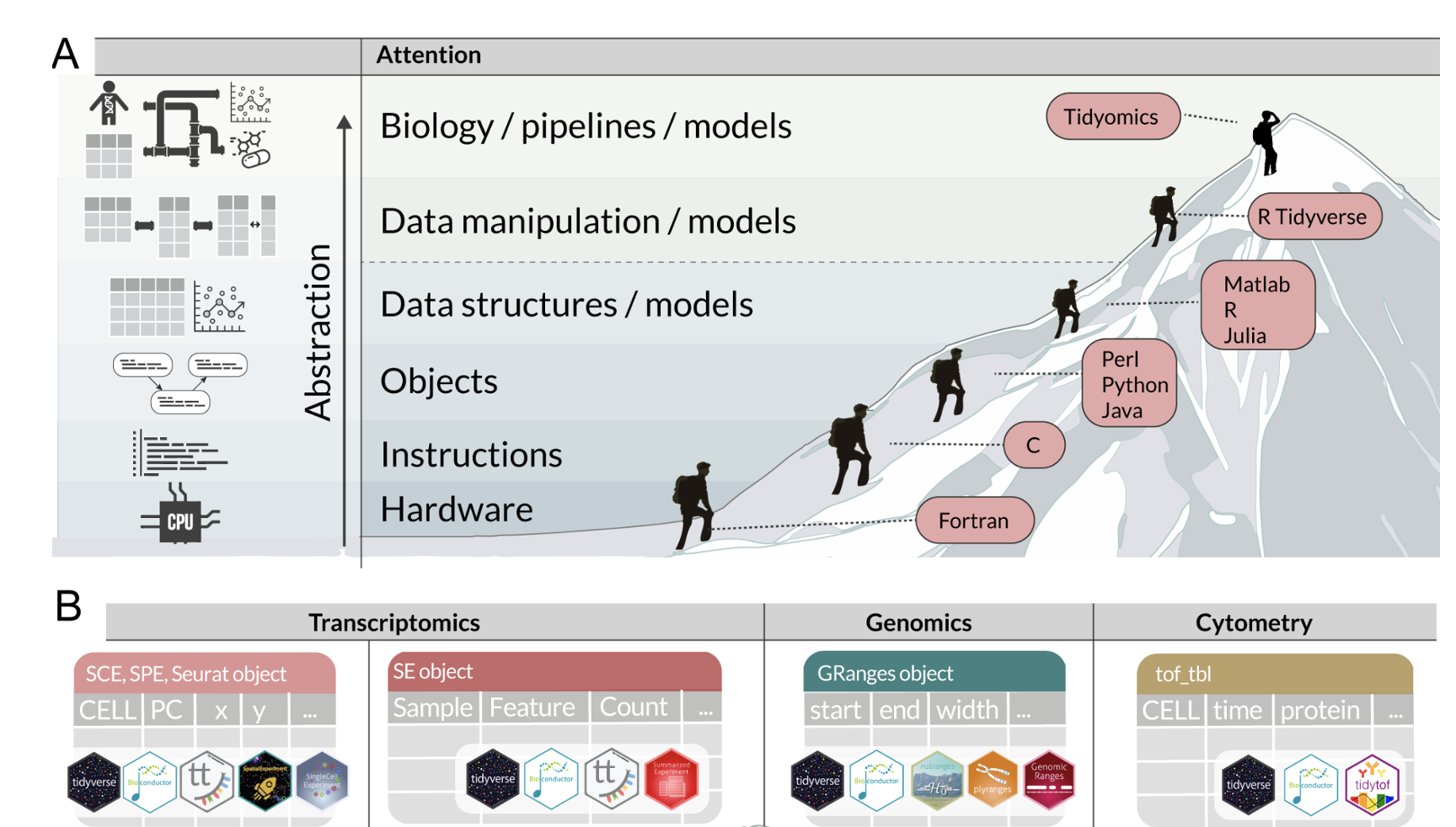
The tidyomics ecosystem: Enhancing omic data analyses
The exponential growth of omic data presents challenges in data manipulation, analysis, and integration. Addressing these challenges, Bioconductor offers an extensive data analysis platform and community, while R tidy programming offers a standard data organisation and manipulation that has revolutionised data science. Bioconductor and tidy R have mostly remained independent; bridging these two ecosystems would streamline omic analysis, ease learning and encourage cross-disciplinary collaborations. Here, we introduce the tidyomics ecosystem—a suite of interoperable software that brings the vast tidy software ecosystem to omic data analysis.
In Nature Methods,
2024
A community challenge to predict clinical outcomes after immune checkpoint blockade in non-small cell lung cancer
Background:Predictive biomarkers of immune checkpoint inhibitor (ICI) efficacy are currently lacking for non-small cell lung cancer (NSCLC). Here, we describe the results from the Anti–PD-1 Response Prediction DREAM Challenge, a crowdsourced initiative that enabled the assessment of predictive models by using data from two randomized controlled clinical trials (RCTs) of ICIs in first-line metastatic NSCLC. Methods Participants developed and trained models using public resources. These were evaluated with data from the CheckMate 026 trial (NCT02041533), according to the model-to-data paradigm to maintain patient confidentiality. The generalizability of the models with the best predictive performance was assessed using data from the CheckMate 227 trial (NCT02477826). Both trials were phase III RCTs with a chemotherapy control arm, which supported the differentiation between predictive and prognostic models. Isolated model containers were evaluated using a bespoke strategy that considered the challenges of handling transcriptome data from clinical trials. Results A total of 59 teams participated, with 417 models submitted. Multiple predictive models, as opposed to a prognostic model, were generated for predicting overall survival, progression-free survival, and progressive disease status with ICIs. Variables within the models submitted by participants included tumor mutational burden (TMB), programmed death ligand 1 (PD-L1) expression, and gene-expression–based signatures. The best-performing models showed improved predictive power over reference variables, including TMB or PD-L1. Conclusions This DREAM Challenge is the first successful attempt to use protected phase III clinical data for a crowdsourced effort towards generating predictive models for ICI clinical outcomes and could serve as a blueprint for similar efforts in other tumor types and disease states, setting a benchmark for future studies aiming to identify biomarkers predictive of ICI efficacy.
In Journal of Translational Medicine,
2024
Embracing the command line: my unexpected career in computational biology
Embracing the command line: my unexpected career in computational biology. A crash course in bioinformatics put Ming Tommy Tang on a different path.
In Nature,
2023
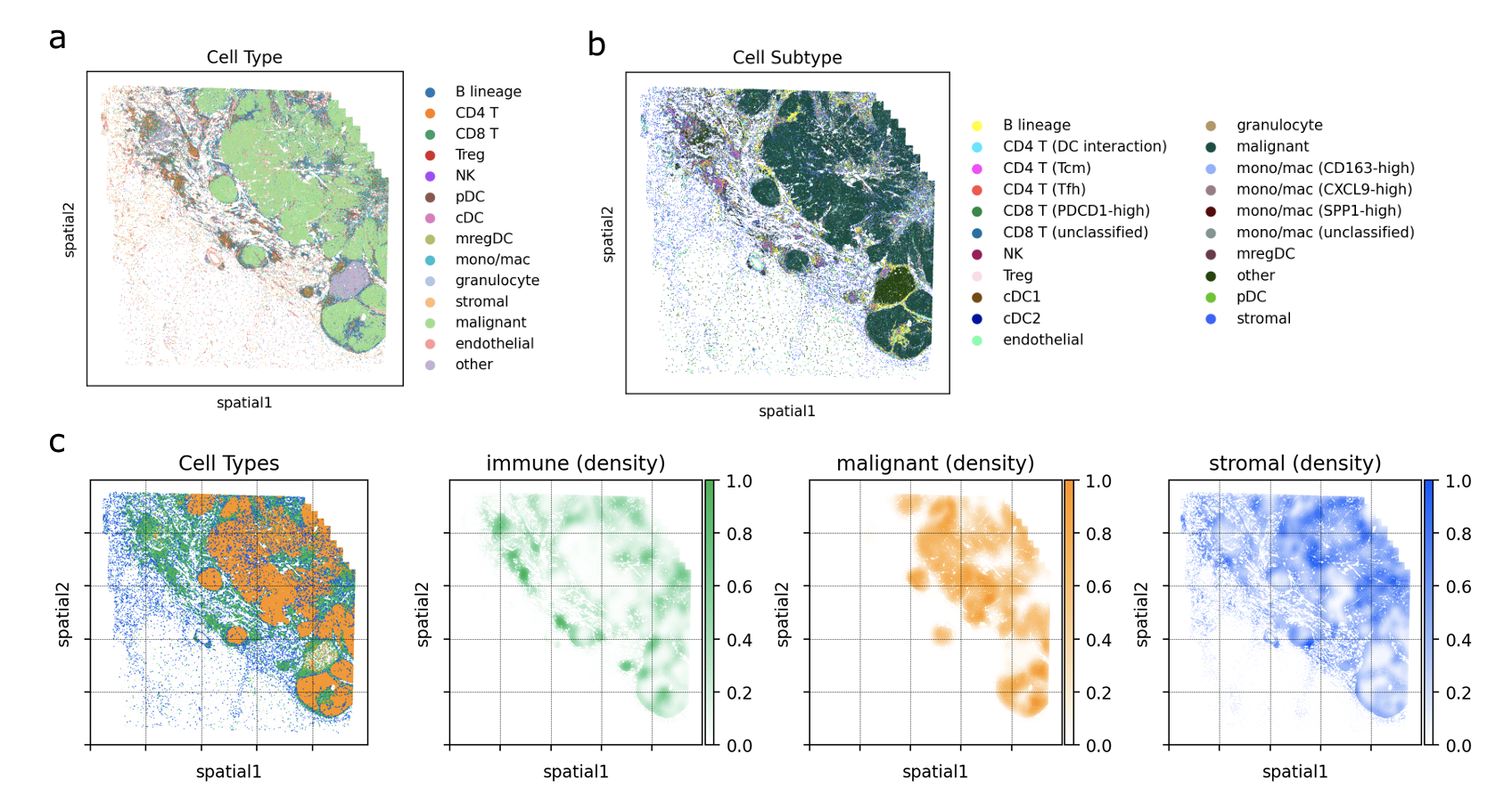
Monkeybread: A Python toolkit for the analysis of cellular niches in single-cell resolution spatial transcriptomics data
Spatial transcriptomics technologies enable the spatially resolved measurement of gene expression within a tissue specimen. With these technologies, researchers can investigate how cells organize into cellular niches which are defined as distinct regions in the tissue comprising a specific composition of cell types or phenotypes. While general-purpose software tools for the exploratory analysis of spatial transcriptomics data exist, there is a need for tools that specialize in the analysis of cellular organization into niches. This can further enhance the downstream application of these data towards drug target discovery, target validation, and biomarker development. We present Monkeybread: A Python toolkit for analyzing cellular organization and intercellular communication in single-cell resolution spatial transcriptomics data. We applied Monkeybread to a human melanoma sample to demonstrate its utility in identifying cellular niches with diverse immunogenic compositions in the tumor microenvironment. We found that these niches were differentially enriched for immunogenic and tolerogenic macrophage populations that could be correlated to T cell abundance. These findings highlight how Monkeybread can be used for revealing underlying biology of the tumor microenvironment, and in the future, for understanding the influence of these niches on response to available treatments and discovery of novel drug targets.
In BioRxiv,
2023
Tutorial: integrative computational analysis of bulk RNA-sequencing data to characterize tumor immunity using RIMA
RNA-sequencing (RNA-seq) has become an increasingly cost-effective technique for molecular profiling and immune characterization of tumors. In the past decade, many computational tools have been developed to characterize tumor immunity from gene expression data. However, the analysis of large-scale RNA-seq data requires bioinformatics proficiency, large computational resources and cancer genomics and immunology knowledge. In this tutorial, we provide an overview of computational analysis of bulk RNA-seq data for immune characterization of tumors and introduce commonly used computational tools with relevance to cancer immunology and immunotherapy. These tools have diverse functions such as evaluation of expression signatures, estimation of immune infiltration, inference of the immune repertoire, prediction of immunotherapy response, neoantigen detection and microbiome quantification. We describe the RNA-seq IMmune Analysis (RIMA) pipeline integrating many of these tools to streamline RNA-seq analysis. We also developed a comprehensive and user-friendly guide in the form of a GitBook with text and video demos to assist users in analyzing bulk RNA-seq data for immune characterization at both individual sample and cohort levels by using RIMA
In Nature Protocols,
2023
Comprehensive Characterizations of Immune Receptor Repertoire in Tumors and Cancer Immunotherapy Studies
We applied our computational algorithm TRUST4 to assemble immune receptor (T-cell receptor/B-cell receptor) repertoires from approximately 12,000 RNA sequencing samples from The Cancer Genome Atlas and seven immunotherapy studies. From over 35 million assembled complete complementary-determining region 3 sequences, we observed that the expression of CCL5 and MZB1 is the most positively correlated genes with T-cell clonal expansion and B-cell clonal expansion, respectively. We analyzed amino acid evolution during B-cell receptor somatic hypermutation and identified tyrosine as the preferred residue. We found that IgG1+IgG3 antibodies together with FcRn were associated with complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity or phagocytosis. In addition to B-cell infiltration, we discovered that B-cell clonal expansion and IgG1+IgG3 antibodies are also correlated with better patient outcomes. Finally, we created a website, VisualizIRR, for users to interactively explore and visualize the immune repertoires in this study.
In Cancer Immunology Research,
2022
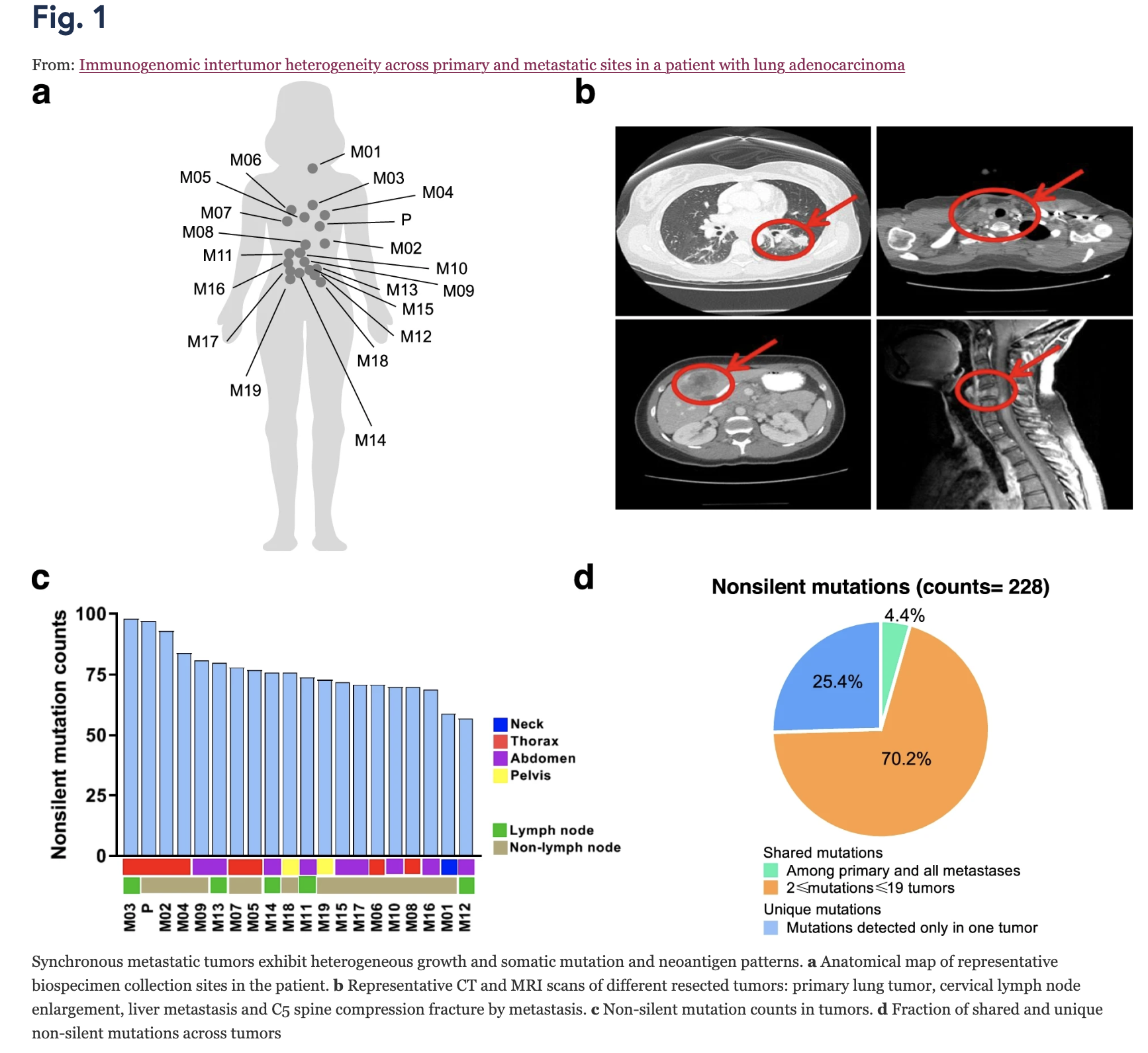
Immunogenomic intertumor heterogeneity across primary and metastatic sites in a patient with lung adenocarcinoma
Background Lung cancer is the leading cause of cancer death, partially owing to its extensive heterogeneity. The analysis of intertumor heterogeneity has been limited by an inability to concurrently obtain tissue from synchronous metastases unaltered by multiple prior lines of therapy. Methods In order to study the relationship between genomic, epigenomic and T cell repertoire heterogeneity in a rare autopsy case from a 32-year-old female never-smoker with left lung primary late-stage lung adenocarcinoma (LUAD), we did whole-exome sequencing (WES), DNA methylation and T cell receptor (TCR) sequencing to characterize the immunogenomic landscape of one primary and 19 synchronous metastatic tumors. Results We observed heterogeneous mutation, methylation, and T cell patterns across distinct metastases. Only TP53 mutation was detected in all tumors suggesting an early event while other cancer gene mutations were later events which may have followed subclonal diversification. A set of prevalent T cell clonotypes were completely excluded from left-side thoracic tumors indicating distinct T cell repertoire profiles between left-side and non left-side thoracic tumors. Though a limited number of predicted neoantigens were shared, these were associated with homology of the T cell repertoire across metastases. Lastly, ratio of methylated neoantigen coding mutations was negatively associated with T-cell density, richness and clonality, suggesting neoantigen methylation may partially drive immunosuppression. Conclusions Our study demonstrates heterogeneous genomic and T cell profiles across synchronous metastases and how restriction of unique T cell clonotypes within an individual may differentially shape the genomic and epigenomic landscapes of synchronous lung metastases.
In Journal of Experimental & Clinical Cancer Research,
2022
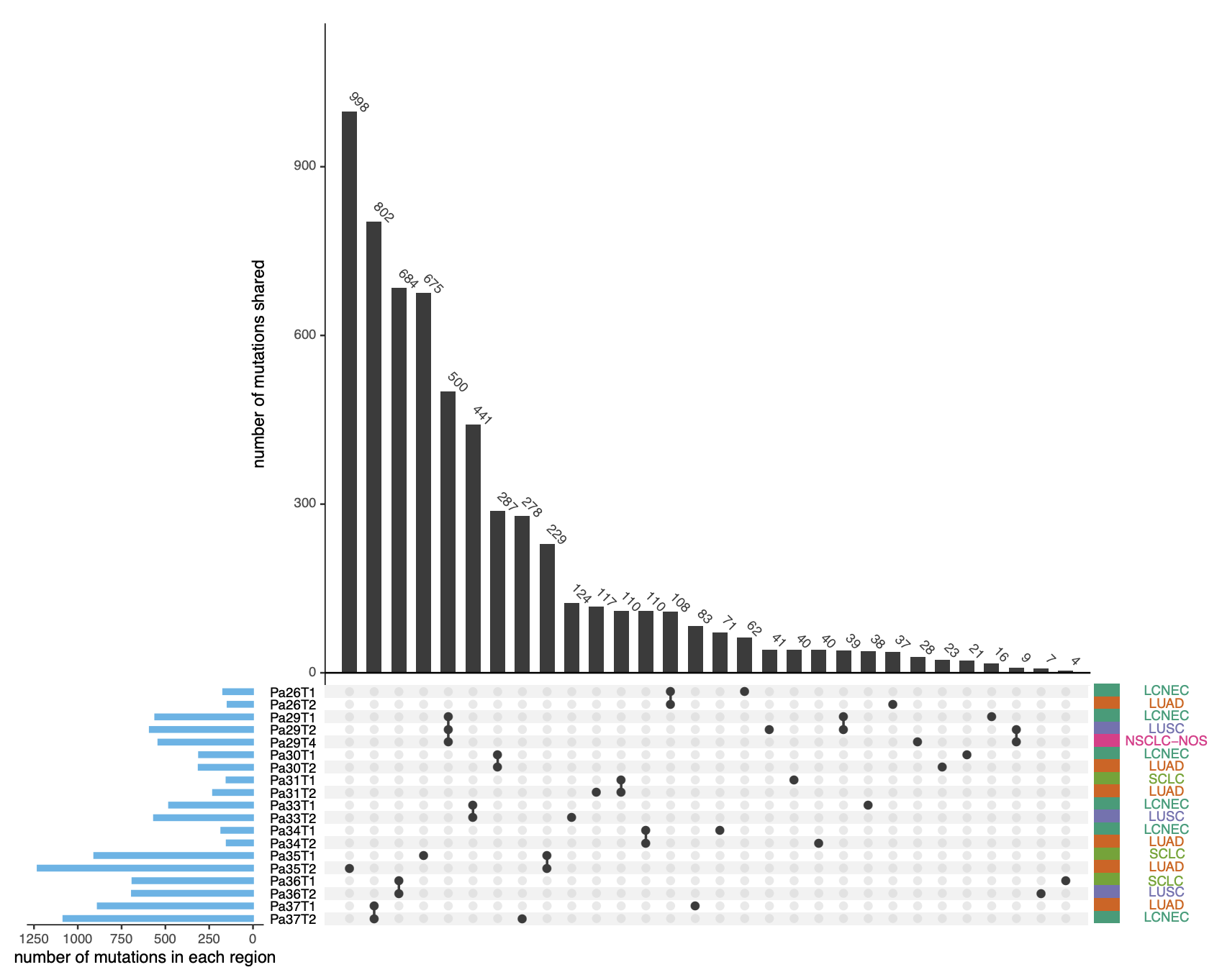
The histologic phenotype of lung cancers may be driven by transcriptomic features rather than genomic characteristics
Histology plays an essential role in therapeutic decision-making for lung cancer patients. However, the molecular determinants of lung cancer histology are largely unknown. We conduct whole-exome sequencing and microarray profiling on 19 micro-dissected tumor regions of different histologic subtypes from 9 patients with lung cancers of mixed histology. A median of 68.9% of point mutations and 83% of copy number aberrations are shared between different histologic components within the same tumors. Furthermore, different histologic components within the tumors demonstrate similar subclonal architecture. On the other hand, transcriptomic profiling reveals shared pathways between the same histologic subtypes from different patients, which is supported by the analyses of the transcriptomic data from 141 cell lines and 343 lung cancers of different histologic subtypes. These data derived from mixed histologic subtypes in the setting of identical genetic background and exposure history support that the histologic fate of lung cancer cells is associated with transcriptomic features rather than the genomic profiles in most tumors..
in Nature Communications,
2021
Fast alignment and preprocessing of chromatin profiles with Chromap
As sequencing depth of chromatin studies continually grows deeper for sensitive profiling of regulatory elements or chromatin spatial structures, aligning and preprocessing of these sequencing data have become the bottleneck for analysis. Here we present Chromap, an ultrafast method for aligning and preprocessing high throughput chromatin profiles. Chromap is comparable to BWA-MEM and Bowtie2 in alignment accuracy and is over 10 times faster than traditional workflows on bulk ChIP-seq/Hi-C profiles and than 10x Genomics’ CellRanger v2.0.0 pipeline on single-cell ATAC-seq profiles.
In Nature Communications,
2021
Single-cell multimodal glioma analyses identify epigenetic regulators of cellular plasticity and environmental stress response
Glioma intratumoral heterogeneity enables adaptation to challenging microenvironments and contributes to therapeutic resistance. We integrated 914 single-cell DNA methylomes, 55,284 single-cell transcriptomes and bulk multi-omic profiles across 11 adult IDH mutant or IDH wild-type gliomas to delineate sources of intratumoral heterogeneity. We showed that local DNA methylation disorder is associated with cell-cell DNA methylation differences, is elevated in more aggressive tumors, links with transcriptional disruption and is altered during the environmental stress response. Glioma cells under in vitro hypoxic and irradiation stress increased local DNA methylation disorder and shifted cell states. We identified a positive association between genetic and epigenetic instability that was supported in bulk longitudinally collected DNA methylation data. Increased DNA methylation disorder associated with accelerated disease progression and recurrently selected DNA methylation changes were enriched for environmental stress response pathways. Our work identified an epigenetically facilitated adaptive stress response process and highlights the importance of epigenetic heterogeneity in shaping therapeutic outcomes.
In Nature Genetics,
2021
Enhancer reprogramming in PRC2-deficient malignant peripheral nerve sheath tumors induces a targetable de-differentiated state
Malignant peripheral nerve sheath tumors (MPNSTs) are soft tissue sarcomas that frequently harbor genetic alterations in polycomb repressor complex 2 (PRC2) components-SUZ12 and EED. Here, we show that PRC2 loss confers a dedifferentiated early neural-crest phenotype which is exclusive to PRC2-mutant MPNSTs and not a feature of neurofibromas. Neural crest phenotype in PRC2 mutant MPNSTs was validated via cross-species comparative analysis using spontaneous and transgenic MPNST models. Systematic chromatin state profiling of the MPNST cells showed extensive epigenomic reprogramming or chromatin states associated with PRC2 loss and identified gains of active enhancer states/super-enhancers on early neural crest regulators in PRC2-mutant conditions around genomic loci that harbored repressed/poised states in PRC2-WT MPNST cells. Consistently, inverse correlation between H3K27me3 loss and H3K27Ac gain was noted in MPNSTs. Epigenetic editing experiments established functional roles for enhancer gains on DLX5-a key regulator of neural crest phenotype. Consistently, blockade of enhancer activity by bromodomain inhibitors specifically suppressed this neural crest phenotype and tumor burden in PRC2-mutant PDXs. Together, these findings reveal accumulation of dedifferentiated neural crest like state in PRC2-mutant MPNSTs that can be targeted by enhancer blockade.
In Acta Neuropathol,
2021
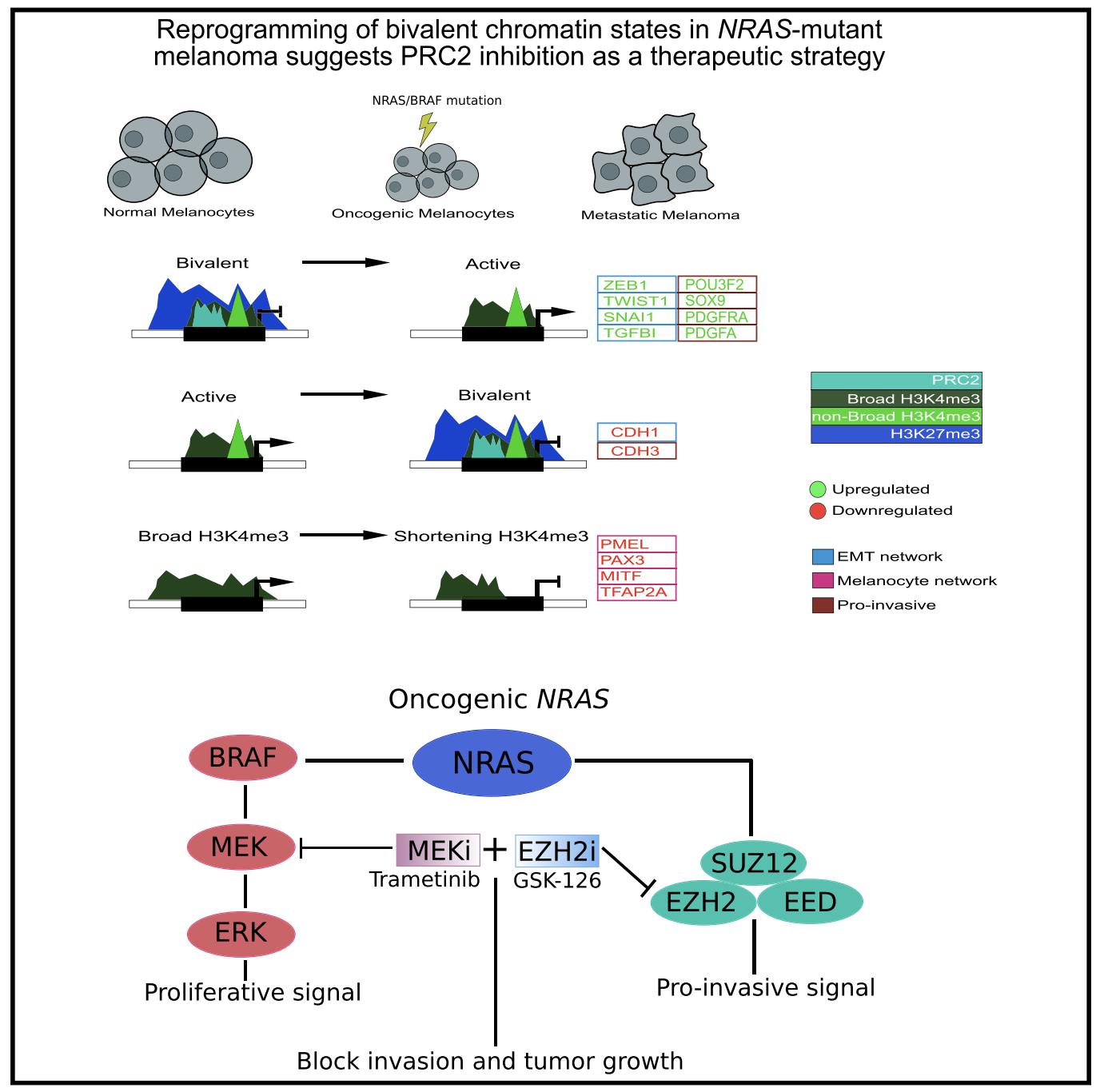
Reprogramming of bivalent chromatin states in NRAS mutant melanoma suggests PRC2 inhibition as a therapeutic strategy
The dynamic evolution of chromatin state patterns during metastasis, their relationship with bona fide genetic drivers, and their therapeutic vulnerabilities are not completely understood. Combinatorial chromatin state profiling of 46 melanoma samples reveals an association of NRAS mutants with bivalent histone H3 lysine 27 trimethylation (H3K27me3) and Polycomb repressive complex 2. Reprogramming of bivalent domains during metastasis occurs on master transcription factors of a mesenchymal phenotype, including ZEB1, TWIST1, and CDH1. Resolution of bivalency using pharmacological inhibition of EZH2 decreases invasive capacity of melanoma cells and markedly reduces tumor burden in vivo, specifically in NRAS mutants. Coincident with bivalent reprogramming, the increased expression of pro-metastatic and melanocyte-specific cell-identity genes is associated with exceptionally wide H3K4me3 domains, suggesting a role for this epigenetic element. Overall, we demonstrate that reprogramming of bivalent and broad domains represents key epigenetic alterations in metastatic melanoma and that EZH2 plus MEK inhibition may provide a promising therapeutic strategy for NRAS mutant melanoma patients.
In Cell Reports,
2021
HieRFIT: A hierarchical cell type classification tool for projections from complex single-cell atlas datasets
Motivation. The emergence of single-cell RNA sequencing (scRNA-seq) has led to an explosion in novel methods to study biological variation among individual cells, and to classify cells into functional and biologically meaningful categories.Results. Here, we present a new cell type projection tool, HieRFIT (Hierarchical Random Forest for Information Transfer), based on hierarchical random forests. HieRFIT uses a priori information about cell type relationships to improve classification accuracy, taking as input a hierarchical tree structure representing the class relationships, along with the reference data. We use an ensemble approach combining multiple random forest models, organized in a hierarchical decision tree structure. We show that our hierarchical classification approach improves accuracy and reduces incorrect predictions especially for inter-dataset tasks which reflect real life applications. We use a scoring scheme that adjusts probability distributions for candidate class labels and resolves uncertainties while avoiding the assignment of cells to incorrect types by labeling cells at internal nodes of the hierarchy when necessary. Availability.HieRFIT is implemented as an R package, and it is available at (https://github.com/yasinkaymaz/HieRFIT/releases/tag/v1.0.0)
In Bioinformatics,
2021
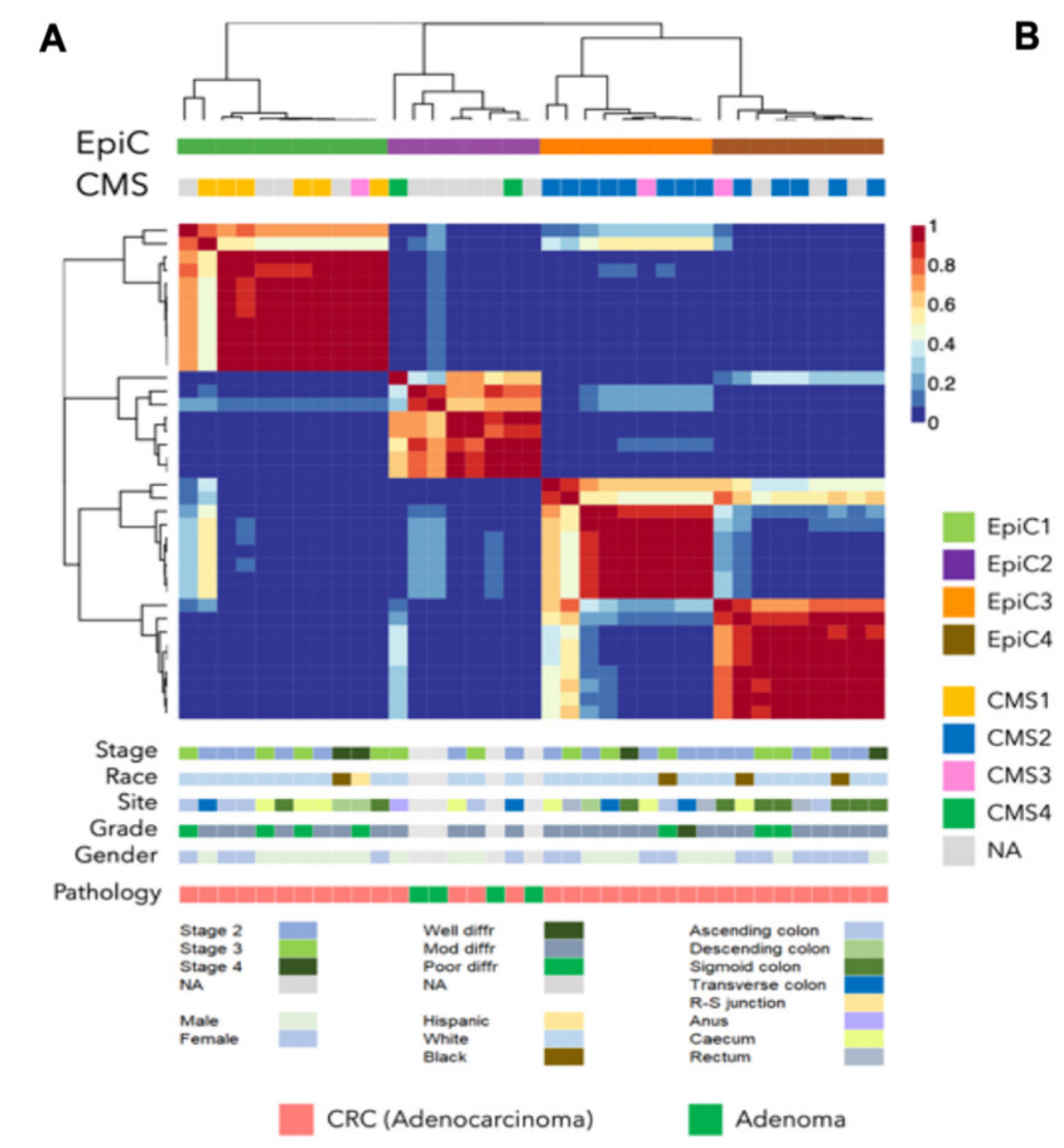
Chromatin state dynamics confers specific therapeutic strategies in enhancer subtypes of colorectal cancer
Objective Enhancer aberrations are beginning to emerge as a key epigenetic feature of colorectal cancers (CRC), however, a comprehensive knowledge of chromatin state patterns in tumour progression, heterogeneity of these patterns and imparted therapeutic opportunities remain poorly described. Design We performed comprehensive epigenomic characterisation by mapping 222 chromatin profiles from 69 samples (33 colorectal adenocarcinomas, 4 adenomas, 21 matched normal tissues and 11 colon cancer cell lines) for six histone modification marks: H3K4me3 for Pol II-bound and CpG-rich promoters, H3K4me1 for poised enhancers, H3K27ac for enhancers and transcriptionally active promoters, H3K79me2 for transcribed regions, H3K27me3 for polycomb repressed regions and H3K9me3 for heterochromatin.Results We demonstrate that H3K27ac-marked active enhancer state could distinguish between different stages of CRC progression. By epigenomic editing, we present evidence that gains of tumour-specific enhancers for crucial oncogenes, such as ASCL2 and FZD10, was required for excessive proliferation. Consistently, combination of MEK plus bromodomain inhibition was found to have synergistic effects in CRC patient-derived xenograft models. Probing intertumour heterogeneity, we identified four distinct enhancer subtypes (EPIgenome-based Classification, EpiC), three of which correlate well with previously defined transcriptomic subtypes (consensus molecular subtypes, CMSs). Importantly, CMS2 can be divided into two EpiC subgroups with significant survival differences. Leveraging such correlation, we devised a combinatorial therapeutic strategy of enhancer-blocking bromodomain inhibitors with pathway-specific inhibitors (PARPi, EGFRi, TGFβi, mTORi and SRCi) for EpiC groups.Conclusion Our data suggest that the dynamics of active enhancer underlies CRC progression and the patient-specific enhancer patterns can be leveraged for precision combination therapy.
In Gut,
2021
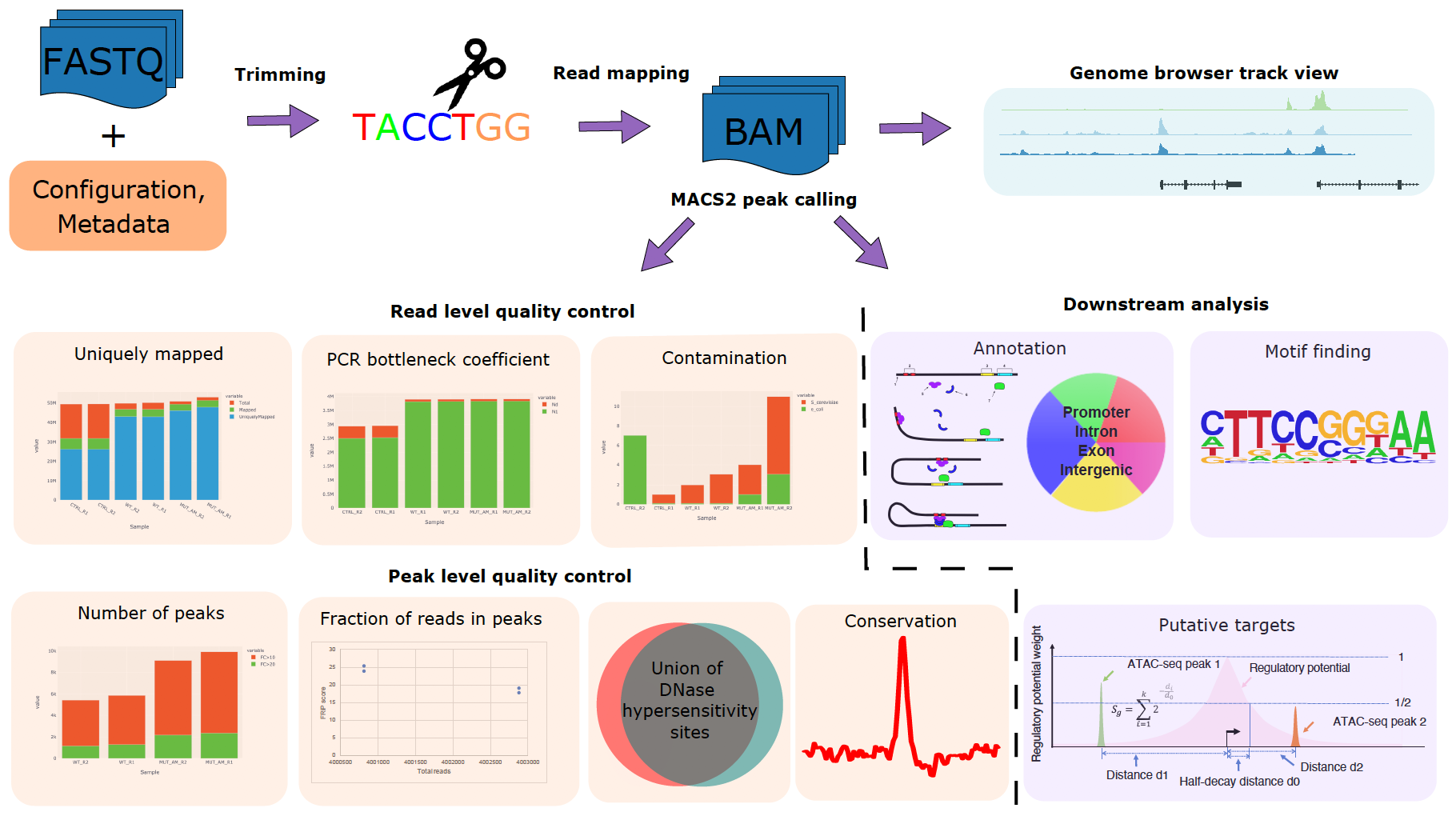
CHIPS: A Snakemake pipeline for quality control and reproducible processing of chromatin profiling data
Motivation: The chromatin profile measured by ATAC-seq, ChIP-seq, or DNase-seq experiments can identify genomic regions critical in regulating gene expression and provide insights on biological processes such as diseases and development. However, quality control and processing chromatin profiling data involve many steps, and different bioinformatics tools are used at each step. It can be challenging to manage the analysis. Results: We developed a Snakemake pipeline called CHIPS (CHromatin enrichment Processor) to streamline the processing of ChIP-seq, ATAC-seq, and DNase-seq data. The pipeline supports single- and paired-end data and is flexible to start with FASTQ or BAM files. It includes basic steps such as read trimming, mapping, and peak calling. In addition, it calculates quality control metrics such as contamination profiles, PCR bottleneck coefficient, the fraction of reads in peaks, percentage of peaks overlapping with the union of public DNaseI hypersensitivity sites, and conservation profile of the peaks. For downstream analysis, it carries out peak annotations, motif finding, and regulatory potential calculation for all genes. The pipeline ensures that the processing is robust and reproducible. Availability: CHIPS is available at https://bitbucket.org/plumbers/cidc_chips/src/master/ Contact: mtang@ds.dfci.harvard.edu; henry_long@dfci.harvard.edu
In F1000Research,
2021
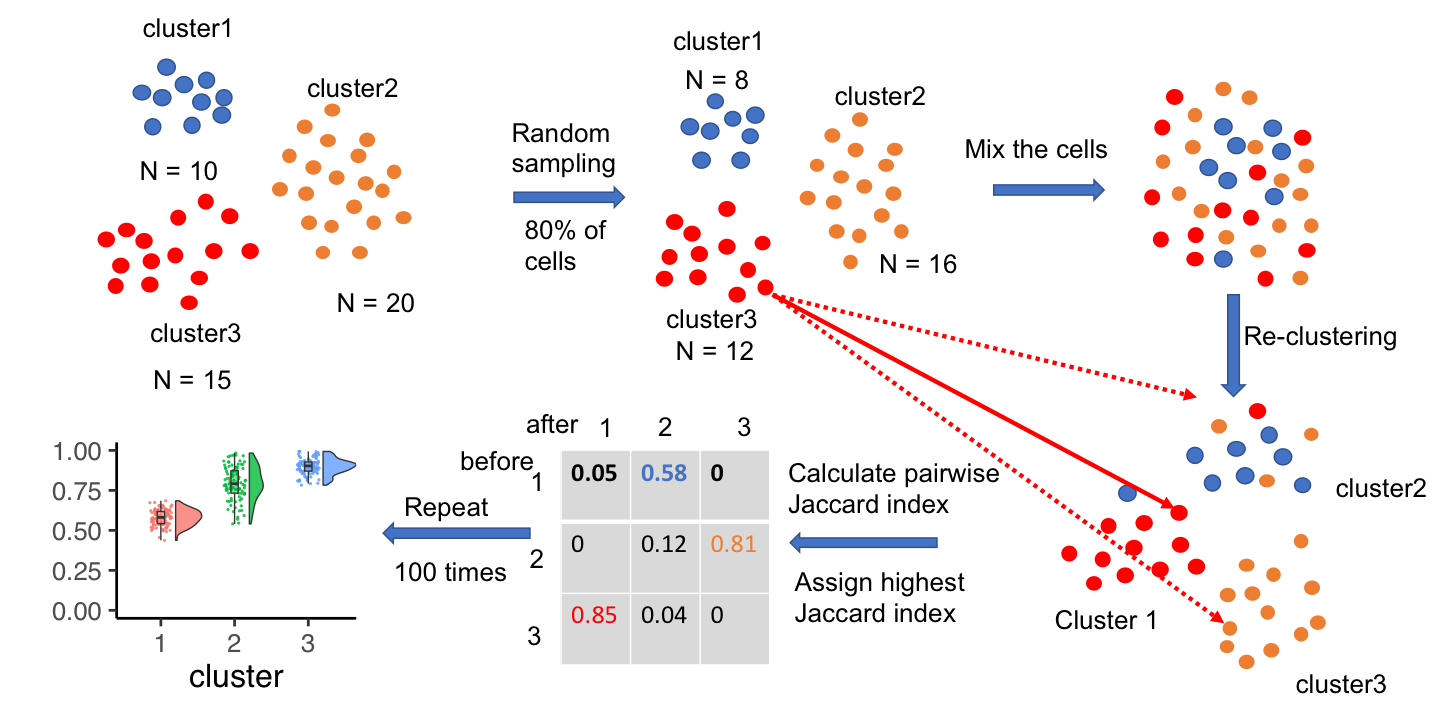
Evaluating single-cell cluster stability using the Jaccard similarity index
Motivation: One major goal of single-cell RNA sequencing (scRNAseq) experiments is to identify novel cell types. With increasingly large scRNAseq datasets, unsupervised clustering methods can now produce detailed catalogues of transcriptionally distinct groups of cells in a sample. However, the interpretation of these clusters is challenging for both technical and biological reasons. Popular clustering algorithms are sensitive to parameter choices, and can produce different clustering solutions with even small changes in the number of principal components used, the k nearest neighbor, and the resolution parameters, among others. Results: Here, we present a set of tools to evaluate cluster stability by subsampling, which can guide parameter choice and aid in biological interpretation. The R package scclusteval and the accompanying Snakemake workflow implement all steps of the pipeline: subsampling the cells, repeating the clustering with Seurat, and estimation of cluster stability using the Jaccard similarity index. The Snakemake workflow takes advantage of high-performance computing clusters and dispatches jobs in parallel to available CPUs to speed up the analysis. The scclusteval package provides functions to facilitate the analysis of the output, including a series of rich visualizations. Availability: R package scclusteval: https://github.com/crazyhottommy/scclusteval Snakemake workflow: https://github.com/crazyhottommy/pyflow_seuratv3_parameter.
In Bioinformatics,
2020
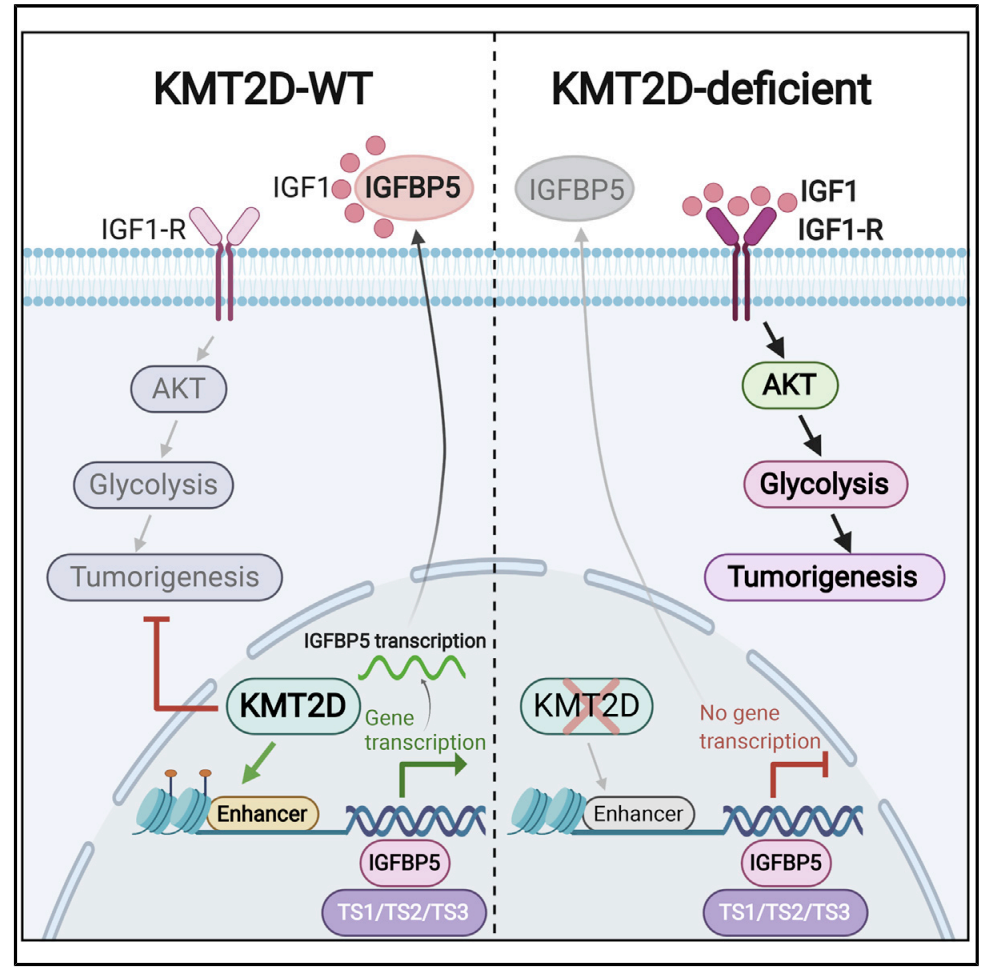
Enhancer Reprogramming Confers Dependence on Glycolysis and IGF Signaling in KMT2D Mutant Melanoma
Histone methyltransferase KMT2D harbors frequent loss-of-function somatic point mutations in several tumor types, including melanoma. Here, we identify KMT2D as a potent tumor suppressor in melanoma through an in vivo epigenome-focused pooled RNAi screen and confirm the finding by using a genetically engineered mouse model (GEMM) based on conditional and melanocyte-specific deletion of KMT2D. KMT2D-deficient tumors show substantial reprogramming of key metabolic pathways, including glycolysis. KMT2D deficiency aberrantly upregulates glycolysis enzymes, intermediate metabolites, and glucose consumption rates. Mechanistically, KMT2D loss causes genome-wide reduction of H3K4me1-marked active enhancer chromatin states. Enhancer loss and subsequent repression of IGFBP5 activates IGF1R-AKT to increase glycolysis in KMT2D-deficient cells. Pharmacological inhibition of glycolysis and insulin growth factor (IGF) signaling reduce proliferation and tumorigenesis preferentially in KMT2D-deficient cells. We conclude that KMT2D loss promotes tumorigenesis by facilitating an increased use of the glycolysis pathway for enhanced biomass needs via enhancer reprogramming, thus presenting an opportunity for therapeutic intervention through glycolysis or IGF pathway inhibitors.
In Cell Reports,
2020
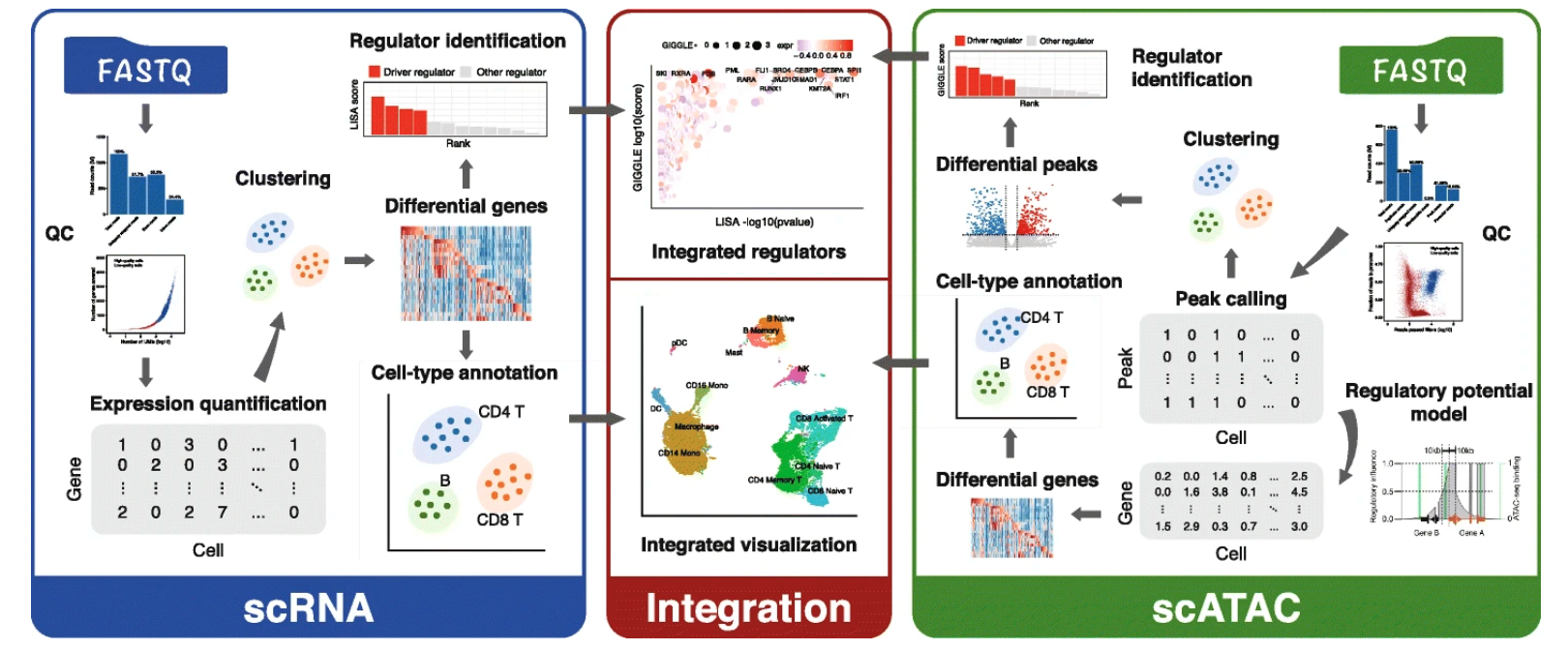
Integrative analyses of single-cell transcriptome and regulome using MAESTRO
We present Model-based AnalysEs of Transcriptome and RegulOme (MAESTRO), a comprehensive open-source computational workflow (http://github.com/liulab-dfci/MAESTRO) for the integrative analyses of single-cell RNA-seq (scRNA-seq) and ATAC-seq (scATAC-seq) data from multiple platforms. MAESTRO provides functions for pre-processing, alignment, quality control, expression and chromatin accessibility quantification, clustering, differential analysis, and annotation. By modeling gene regulatory potential from chromatin accessibilities at the single-cell level, MAESTRO outperforms the existing methods for integrating the cell clusters between scRNA-seq and scATAC-seq. Furthermore, MAESTRO supports automatic cell-type annotation using predefined cell type marker genes and identifies driver regulators from differential scRNA-seq genes and scATAC-seq peaks.
In Genome Biology,
2020
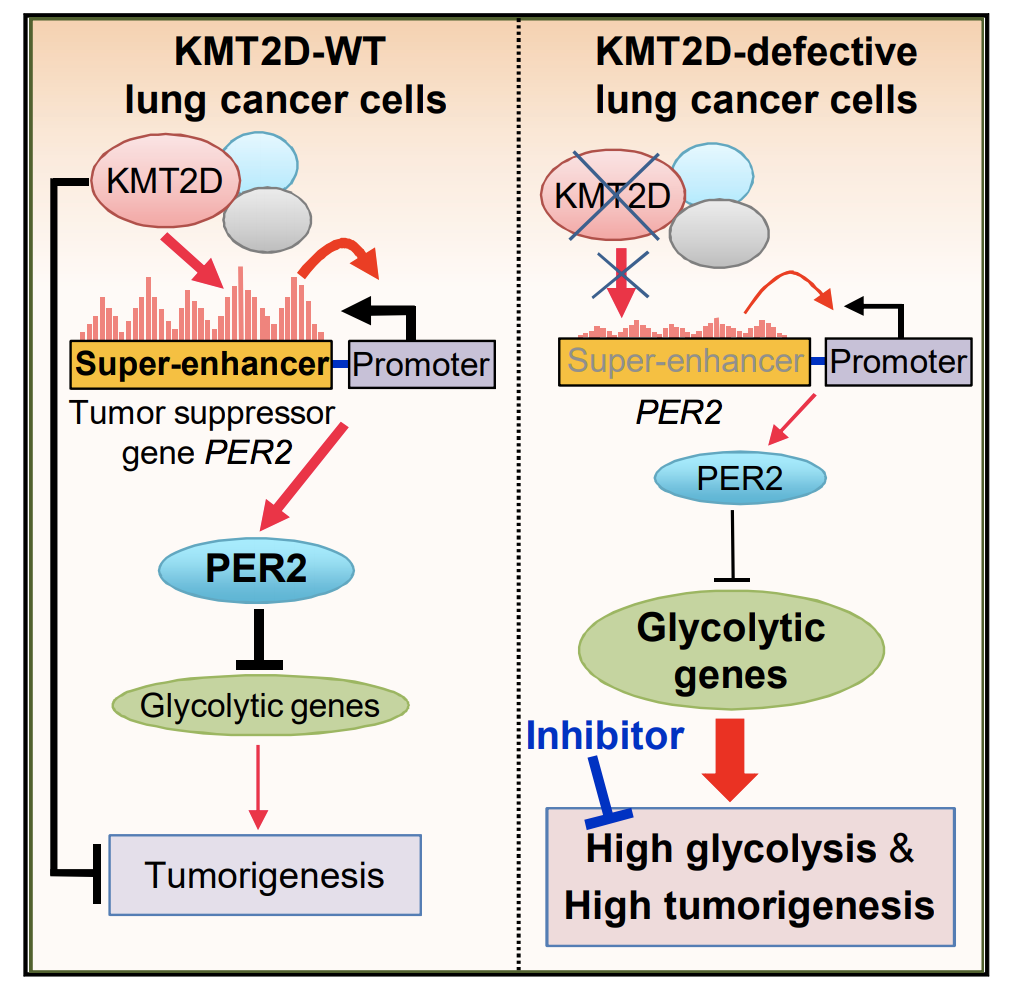
KMT2D Deficiency Impairs Super-Enhancers to Confer a Glycolytic Vulnerability in Lung Cancer
Epigenetic modifiers frequently harbor loss-of-function mutations in lung cancer, but their tumor-suppressive roles are poorly characterized. Histone methyltransferase KMT2D (a COMPASS-like enzyme, also called MLL4) is among the most highly inactivated epigenetic modifiers in lung cancer. Here, we show that lung-specific loss of Kmt2d promotes lung tumorigenesis in mice and upregulates pro-tumorigenic programs, including glycolysis. Pharmacological inhibition of glycolysis preferentially impedes tumorigenicity of human lung cancer cells bearing KMT2D-inactivating mutations. Mechanistically,Kmt2d loss widely impairs epigenomic signals for super-enhancers/enhancers, including the super-enhancer for the circadian rhythm repressor Per2. Loss of Kmt2d decreases expression of PER2, which regulates multiple glycolytic genes. These findings indicate that KMT2D is a lung tumor suppressor and that KMT2D deficiency confers a therapeutic vulnerability to glycolytic inhibitors.
In Cancer Cell, Highlight in ScienceSignaling: Tumor’s loss is clinician’s gain,
2020
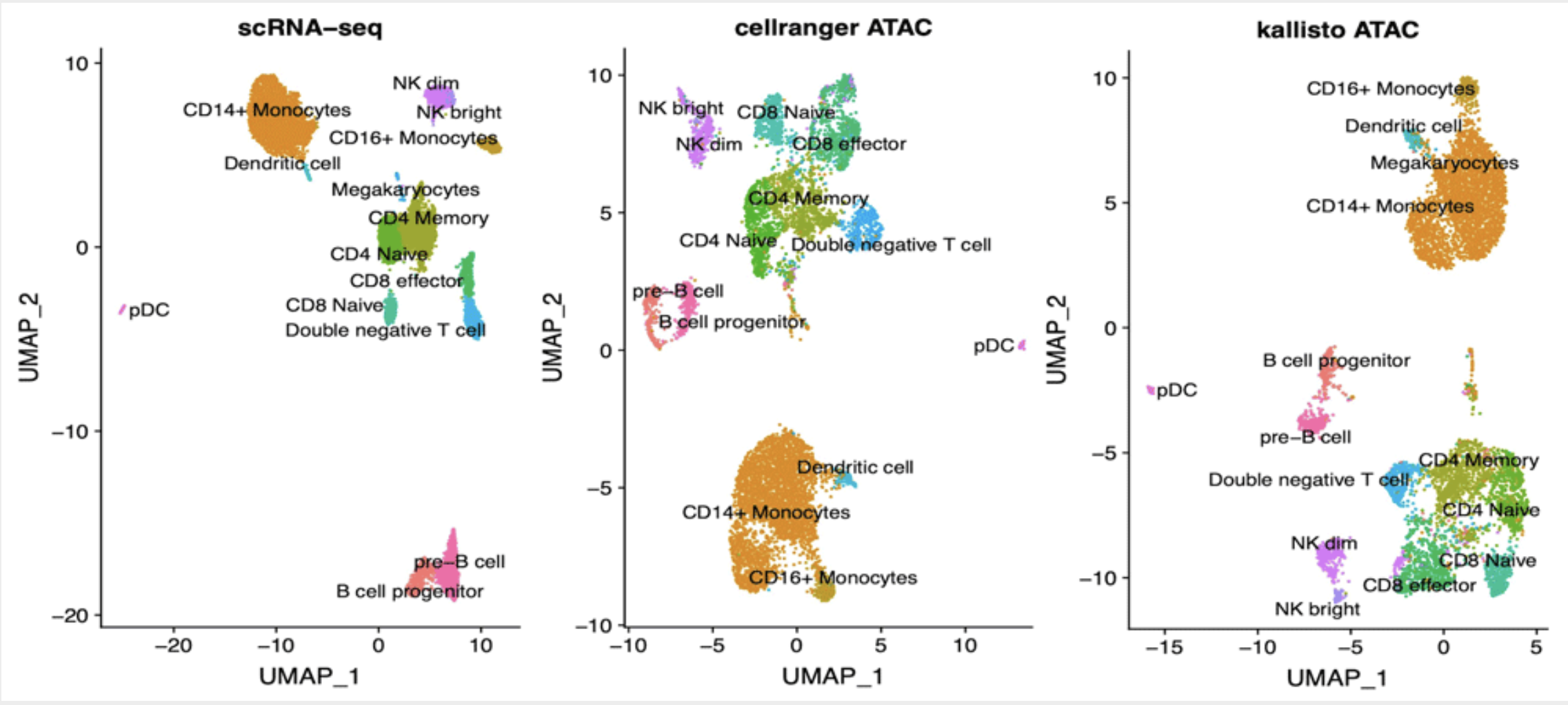
Fast analysis of scATAC-seq data using a predefined set of genomic regions
Background: Analysis of scATAC-seq data has been recently scaled to thousands of cells. While processing of other types of single cell data was boosted by the implementation of alignment-free techniques, pipelines available to process scATAC-seq data still require large computational resources. We propose here an approach based on pseudoalignment, which reduces the execution times and hardware needs at little cost for precision. Methods: Public data for 10k PBMC were downloaded from 10x Genomics web site. Reads were aligned to various references derived from DNase I Hypersensitive Sites (DHS) using kallisto and quantified with bustools. We compared our results with the ones publicly available derived by cellranger-atac. Results: We found that kallisto does not introduce biases in quantification of known peaks and cells groups are identified in a consistent way. We also found that cell identification is robust when analysis is performed using DHS-derived reference in place of de novo identification of ATAC peaks. Lastly, we found that our approach is suitable for reliable quantification of gene activity based on scATAC-seq signal, thus allows for efficient labelling of cell groups based on marker genes.Conclusions: Analysis of scATAC-seq data by means of kallisto produces results in line with standard pipelines while being considerably faster; using a set of known DHS sites as reference does not affect the ability to characterize the cell populations
In F1000 Research,
2020
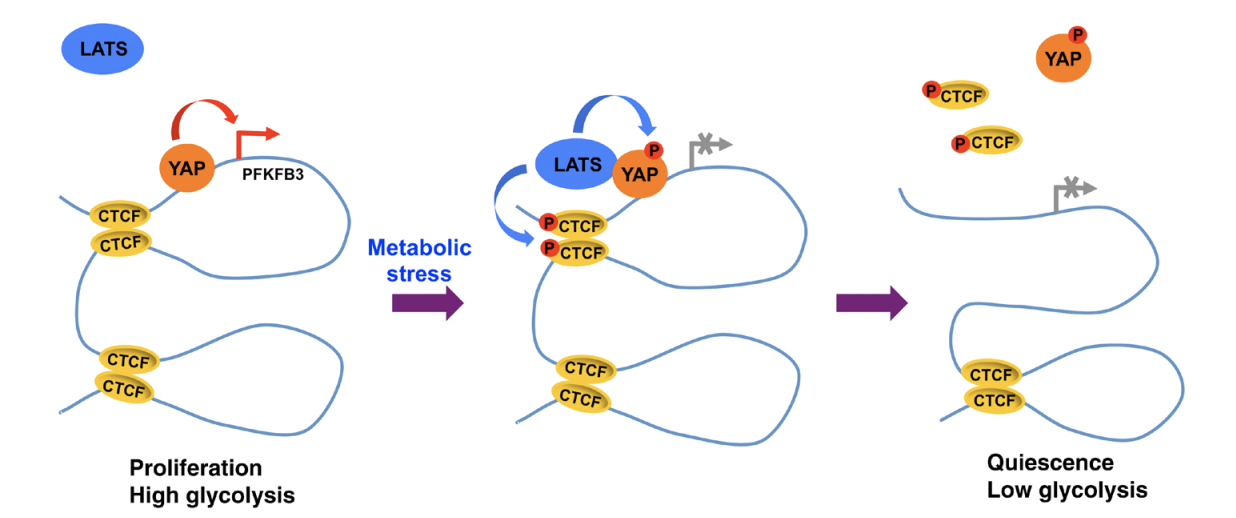
LATS kinase–mediated CTCF phosphorylation and selective loss of genomic binding
Chromatin topological organization is instrumental in gene transcription. Gene-enhancer interactions are accommodated in the same CTCF-mediated insulated neighborhoods. However, it remains poorly understood whether and how the 3D genome architecture is dynamically restructured by external signals. Here, we report that LATS kinases phosphorylated CTCF in the zinc finger (ZF) linkers and disabled its DNA-binding activity. Cellular stress induced LATS nuclear translocation and CTCF ZF linker phosphorylation, and altered the landscape of CTCF genomic binding partly by dissociating it selectively from a small subset of its genomic binding sites. These sites were highly enriched for the boundaries of chromatin domains containing LATS signaling target genes. The stress-induced CTCF phosphorylation and locus-specific dissociation from DNA were LATS-dependent. Loss of CTCF binding disrupted local chromatin domains and down-regulated genes located within them. The study suggests that external signals may rapidly modulate the 3D genome by affecting CTCF genomic binding through ZF linker phosphorylation.
In Science Advances,
2020
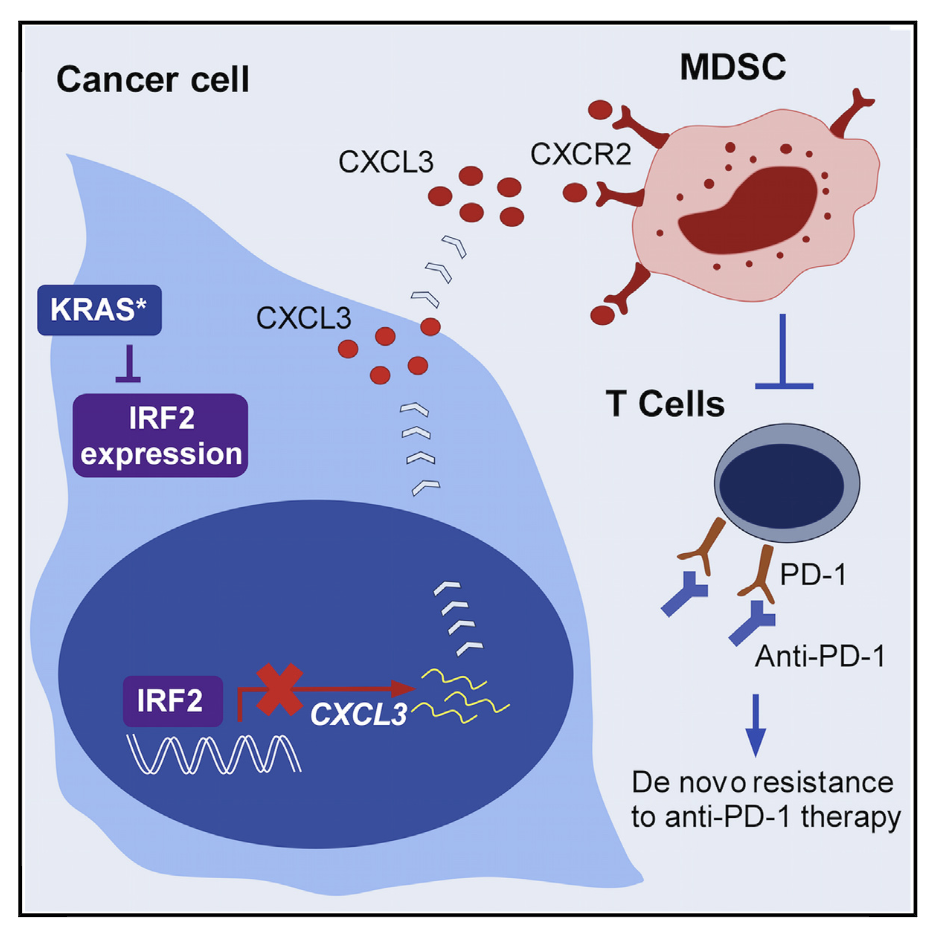
KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer
The biological functions and mechanisms of oncogenic KRASG12D(KRAS) in resistance to immune check-point blockade (ICB) therapy are not fully understood. We demonstrate that KRAS* represses the expressionof interferon regulatory factor 2 (IRF2), which in turn directly represses CXCL3 expression. KRAS*-mediatedrepression of IRF2 results in high expression of CXCL3, which binds to CXCR2 on myeloid-derived suppres-sor cells and promotes their migration to the tumor microenvironment. Anti-PD-1 resistance of KRAS-ex-pressing tumors can be overcome by enforced IRF2 expression or by inhibition of CXCR2. Colorectal cancer(CRC) showing higher IRF2 expression exhibited increased responsiveness to anti-PD-1 therapy. The KRAS-IRF2-CXCL3-CXCR2 axis provides a framework for patient selection and combination therapies to enhancethe effectiveness of ICB therapy in CRC
In Cancer Cell,
2019
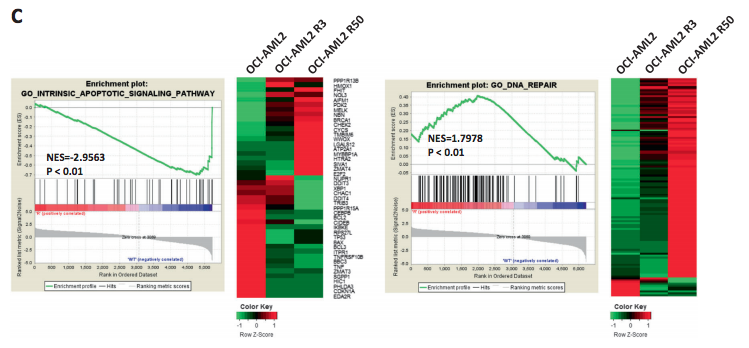
Histone deacetylase inhibitor targets CD123/CD47-positive cells and reverse chemoresistance phenotype in acute myeloid leukemia
Chemoresistance may be due to the survival of leukemia stem cells (LSCs) that are quiescent and not responsive to chemotherapy or lie on the intrinsic or acquired resistance of the specific pool of AML cells. Here, we found, among well-established LSC markers, only CD123 and CD47 are correlated with AML cell chemosensitivities across cell lines and patient samples. Further study reveals that percentages of CD123+CD47+ cells significantly increased in chemoresistant lines compared to parental cell lines. However, stemness signature genes are not significantly increased in resistant cells. Instead, gene changes are enriched in cell cycle and cell survival pathways. This suggests CD123 may serve as a biomarker for chemoresistance, but not stemness of AML cells. We further investigated the role of epigenetic factors in regulating the survival of chemoresistant leukemia cells. Epigenetic drugs, especially histone deacetylase inhibitors (HDACis), effectively induced apoptosis of chemoresistant cells. Furthermore, HDACi Romidepsin largely reversed gene expression profile of resistant cells and efficiently targeted and removed chemoresistant leukemia blasts in xenograft AML mouse model. More interestingly, Romidepsin preferentially targets CD123+ cells, while chemotherapy drug Ara-C mainly targeted fast-growing, CD123− cells. Therefore, Romidepsin alone or in combination with Ara-C may be a potential treatment strategy for chemoresistant patients.
In Leukemia,
2018
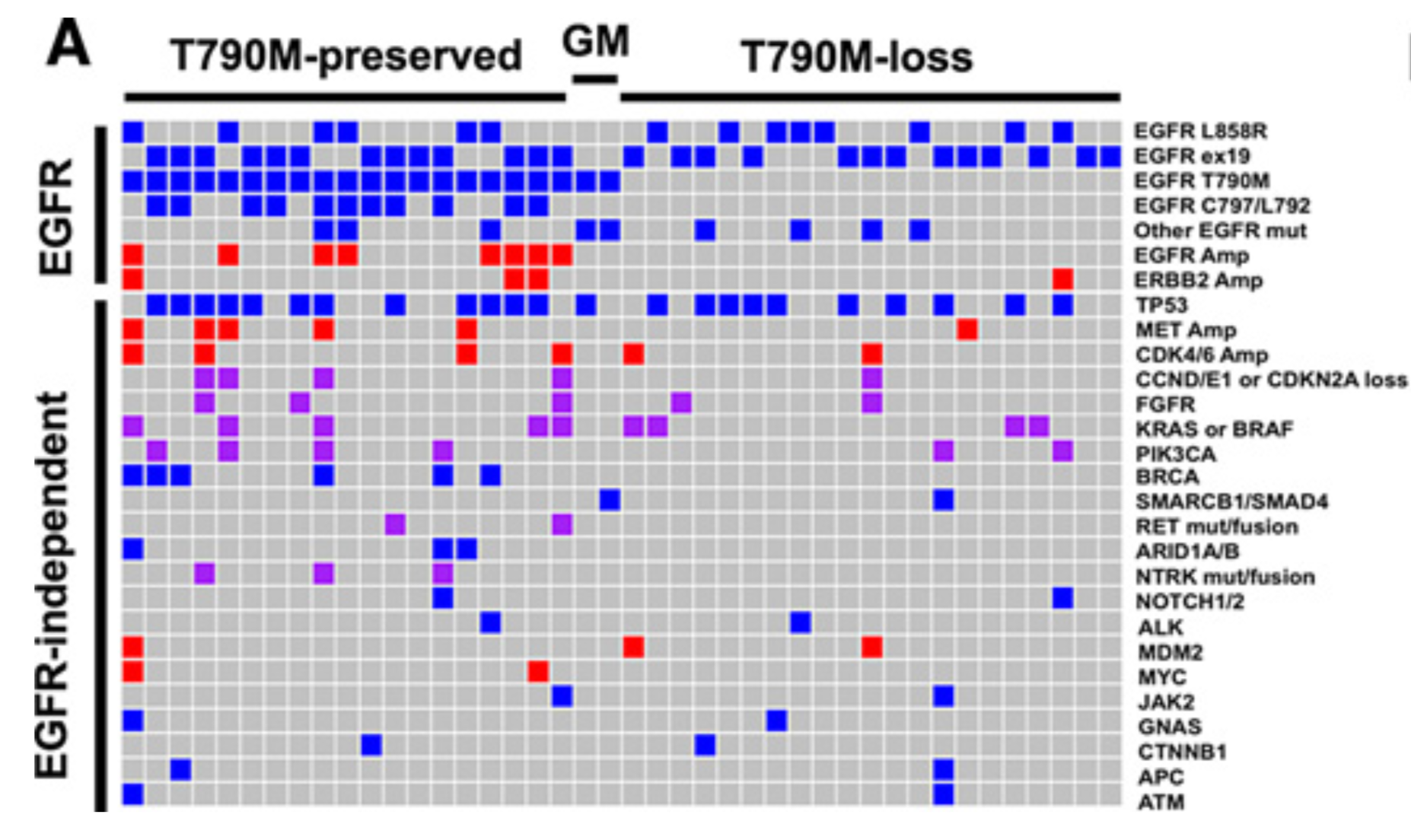
Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC
PURPOSE:Osimertinib was initially approved for T790M-positive non-small cell lung cancer (NSCLC) and, more recently, for first-line treatment of EGFR-mutant NSCLC. However, resistance mechanisms to osimertinib have been incompletely described.EXPERIMENTAL DESIGN:Using cohorts from The University of Texas MD Anderson Lung Cancer Moonshot GEMINI and Moffitt Cancer Center lung cancer databases, we collected clinical data for patients treated with osimertinib. Molecular profiling analysis was performed at the time of progression in a subset of the patients.RESULTS:In the 118 patients treated with osimertinib, 42 had molecular profiling at progression. T790M was preserved in 21 (50%) patients and lost in 21 (50%). EGFR C797 and L792 (26%) mutations were the most common resistance mechanism and were observed exclusively in T790M-preserved cases. MET amplification was the second most common alteration (14%). Recurrent alterations were observed in 22 genes/pathways, including PIK3CA, FGFR, and RET. Preclinical studies confirmed MET, PIK3CA, and epithelial-to-mesenchymal transition as potential resistance drivers. Alterations of cell-cycle genes were associated with shorter median progression-free survival (PFS, 4.4 vs. 8.8 months, P = 0.01). In 76 patients with progression, osimertinib was continued in 47 cases with a median second PFS (PFS2) of 12.6 months; 21 patients received local consolidation radiation with a median PFS of 15.5 months. Continuation of osimertinib beyond progression was associated with a longer overall survival compared with discontinuation (11.2 vs. 6.1 months, P = 0.02). CONCLUSIONS:Osimertinib resistance is associated with diverse, predominantly EGFR-independent genomic alterations. Continuation of osimertinib after progression, alone or in conjunction with radiotherapy, may provide prolonged clinical benefit in selected patients
In Clinical Cancer Research,
2018
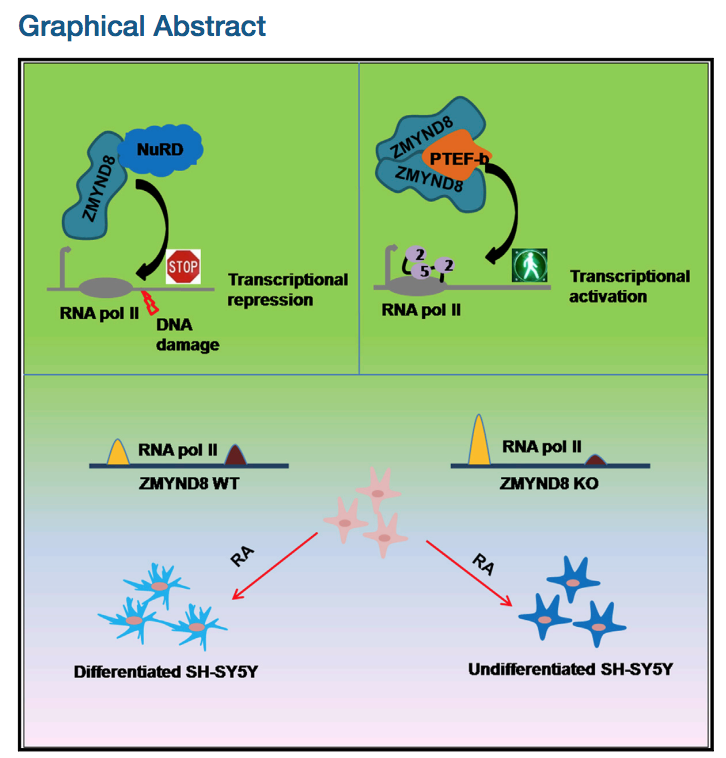
Positive Regulation of Transcription by Human ZMYND8 through Its Association with P-TEFb Complex
Although human ZMYND8 has been implicated as a transcriptional co-repressor of multiple targets, global association of ZMYND8 with active genes and enhancer regions predicts otherwise. Here, we report an additional function of ZMYND8 in transcriptional activation through its association with the P-TEFb complex. Biochemical reconstitution analyses show that human ZMYND8, through direct association with CylcinT1, forms a minimal ZMYND8-P-TEFb complex. The importance of ZMYND8 in target gene activation, through P-TEFb complex recruitment, is demonstrated on chromosomally integrated reporter gene as well as native target genes in vivo. Physiologically, we further show that the ZMYND8-P-TEFb complex-mediated transcriptional activation is required for all-trans retinoic acid (ATRA)-mediated differentiation of neuronal precursor cells. Finally, to detail the dual activator and repressor nature, mechanistically we show that, through its putative coiled-coil domain, ZMYND8 forms a homodimer that preferentially associates with the activator P-TEFb complex, whereas the monomer associates with the CHD4 subunit of repressor NuRD complex.
In Cell Reports,
2018
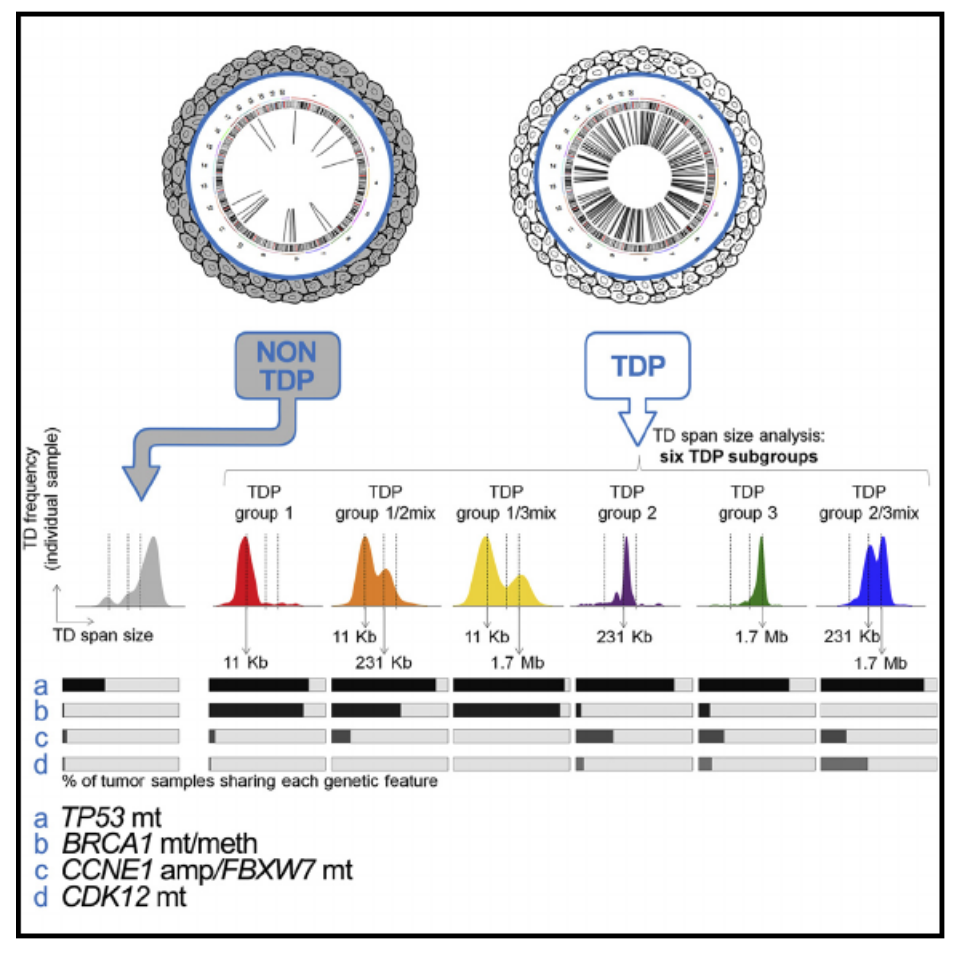
The Tandem Duplicator Phenotype Is a Prevalent Genome-Wide Cancer Configuration Driven by Distinct Gene Mutations
The tandem duplicator phenotype (TDP) is a genome-wide instability configuration primarily observed in breast, ovarian, and endometrial carcinomas. Here, we stratify TDP tumors by classifying their tandem duplications (TDs) into three span intervals, with modal values of 11 kb, 231 kb, and 1.7 Mb, respectively. TDPs with ∼11 kb TDs feature loss of TP53 and BRCA1. TDPs with ∼231 kb and ∼1.7 Mb TDs associate with CCNE1 pathway activation and CDK12 disruptions, respectively. We demonstrate that p53 and BRCA1 conjoint abrogation drives TDP induction by generating short-span TDP mammary tumors in genetically modified mice lacking them. Lastly, we show how TDs in TDP tumors disrupt heterogeneous combinations of tumor suppressors and chromatin topologically associating domains while duplicating oncogenes and super-enhancers.
In Cancer Cell,
2018
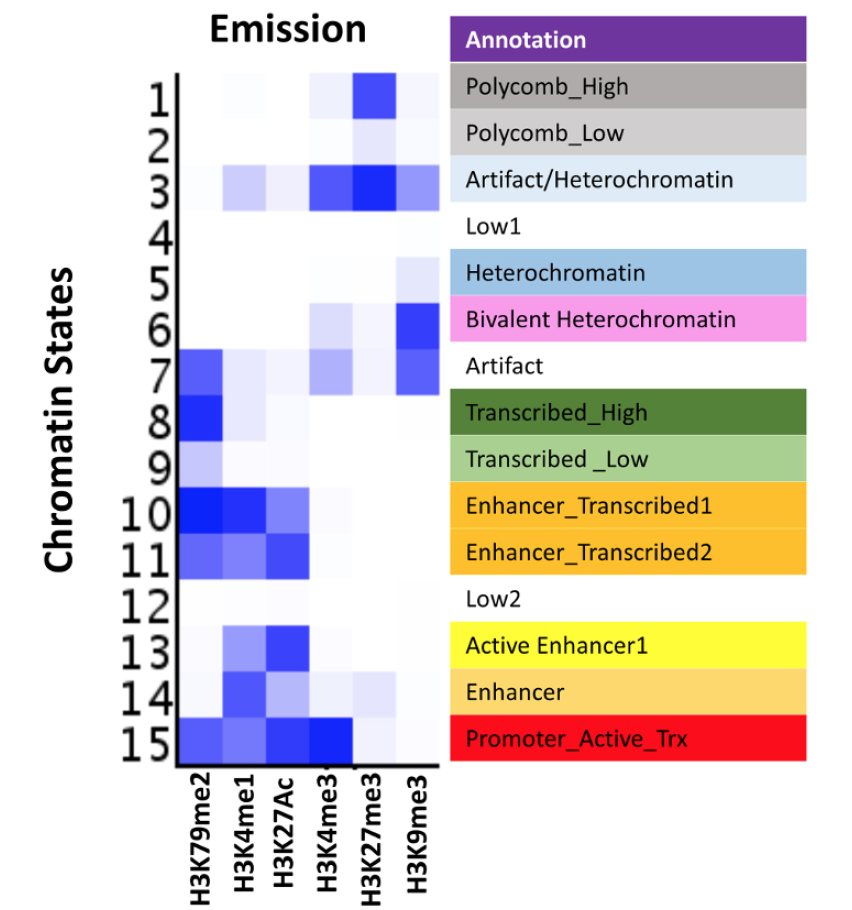
An Integrated Platform for Genome-wide Mapping of Chromatin States Using High-throughput ChIP-sequencing in Tumor Tissues
Histone modifications constitute a major component of the epigenome and play important regulatory roles in determining the transcriptional status of associated loci. In addition, the presence of specific modifications has been used to determine the position and identity non-coding functional elements such as enhancers. In recent years, chromatin immunoprecipitation followed by next generation sequencing (ChIP-seq) has become a powerful tool in determining the genome-wide profiles of individual histone modifications. However, it has become increasingly clear that the combinatorial patterns of chromatin modifications, referred to as Chromatin States, determine the identity and nature of the associated genomic locus. Therefore, workflows consisting of robust high-throughput (HT) methodologies for profiling a number of histone modification marks, as well as computational analyses pipelines capable of handling myriads of ChIP-Seq profiling datasets, are needed for comprehensive determination of epigenomic states in large number of samples. The HT-ChIP-Seq workflow presented here consists of two modules: 1) an experimental protocol for profiling several histone modifications from small amounts of tumor samples and cell lines in a 96-well format; and 2) a computational data analysis pipeline that combines existing tools to compute both individual mark occupancy and combinatorial chromatin state patterns. Together, these two modules facilitate easy processing of hundreds of ChIP-Seq samples in a fast and efficient manner. The workflow presented here is used to derive chromatin state patterns from 6 histone mark profiles in melanoma tumors and cell lines. Overall, we present a comprehensive ChIP-seq workflow that can be applied to dozens of human tumor samples and cancer cell lines to determine epigenomic aberrations in various malignancies.
In JOVE,
2018
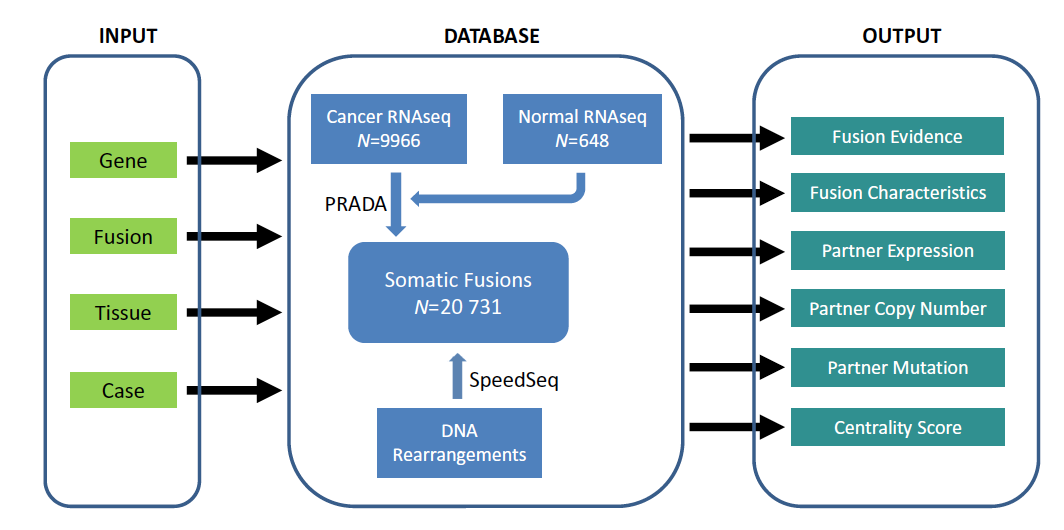
TumorFusions: an integrative resource for cancer-associated transcript fusions
Gene fusion represents a class of molecular aberrations in cancer and has been exploited for therapeutic purposes. In this paper we describe Tumor-Fusions, a data portal that catalogues 20731 gene fusions detected in 9966 well characterized cancer samples and 648 normal specimens from The Cancer Genome Atlas (TCGA). The portal spans 33 cancer types in TCGA. Fusion transcripts were identified via a uniform pipeline, including filtering against a list of 3838 transcript fusions detected in a panel of 648 non-neoplastic samples. Fusions were mapped to somatic DNA rearrangements identified using whole genome sequencing data from 561 cancer samples as a means of validation. We observed that 65% of transcript fusions were associated with a chromosomal alteration, which is annotated in the portal. Other features of the portal include links to SNP array-based copy number levels and mutational patterns, exon and transcript level expressions of the partner genes, and a network-based centrality score for prioritizing functional fusions. Our portal aims to be a broadly applicable and user friendly resource for cancer gene annotation and is publicly available at http://www.tumorfusions.org
In Nucleic Acids Research,
2018
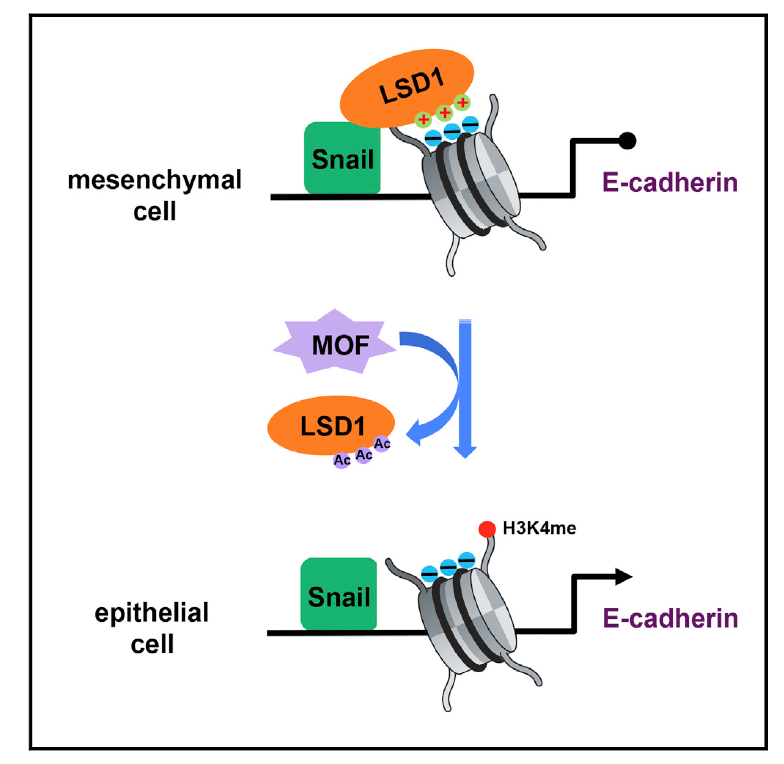
MOF Acetylates the Histone Demethylase LSD1 to Suppress Epithelial-to-Mesenchymal Transition
The histone demethylase LSD1 facilitates epithelialto-mesenchymal transition (EMT) and tumor progression by repressing epithelial marker expression. However, little is known about how its function may be modulated. Here, we report that LSD1 is acetylated in epithelial but not mesenchymal cells. Acetylation of LSD1 reduces its association with nucleosomes, thus increasing histone H3K4 methylation at its target genes and activating transcription. The MOF acetyltransferase interacts with LSD1 and is responsible for its acetylation. MOF is preferentially expressed in epithelial cells and is downregulated by EMT-inducing signals. Expression of exogenous MOF impedes LSD1 binding to epithelial gene promoters and histone demethylation, thereby suppressing EMT and tumor invasion. Conversely, MOFdepletion enhances EMT and tumor metastasis. In human cancer, high MOF expression correlates with epithelial markers and a favorable prognosis. These findings provide insight into the regulation of LSD1 and EMT and identify MOF as a critical suppressor of EMT and tumor progression.
In Cell Reports,
2017
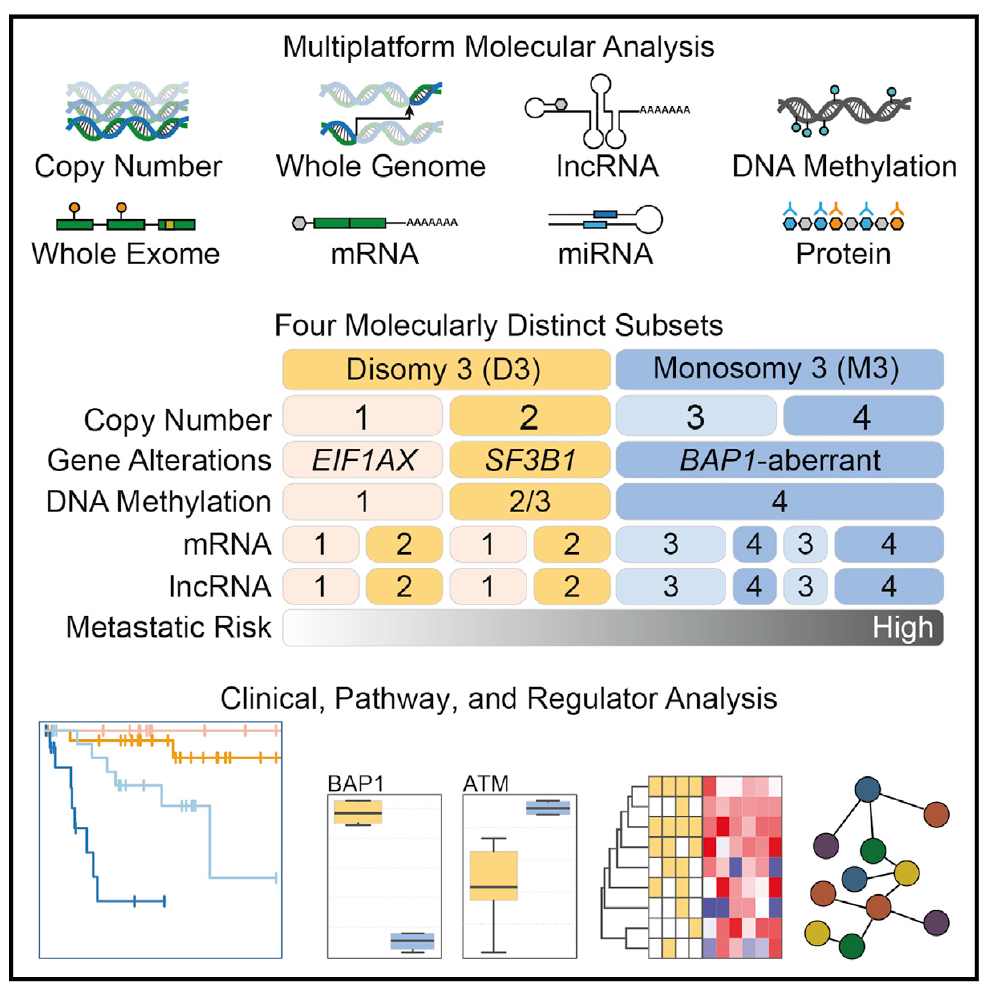
Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma
Comprehensive multiplatform analysis of 80 uveal melanomas (UM) identifies four molecularly distinct, clinically relevant subtypes: two associated with poor-prognosis monosomy 3 (M3) and two with betterprognosis disomy 3 (D3). We show that BAP1 loss follows M3 occurrence and correlates with a global DNA methylation state that is distinct from D3-UM. Poor-prognosis M3-UMdivide into subsets with divergent genomic aberrations, transcriptional features, and clinical outcomes. We report change-of-function SRSF2 mutations. Within D3-UM, EIF1AX- and SRSF2/SF3B1-mutant tumors have distinct somatic copy number alterations and DNA methylation profiles, providing insight into the biology of these low- versus intermediate-risk clinical mutation subtypes.
In Cancer Cell,
2017
ChIP-seq analysis Book chapter for Biostar Handbook
In 2017, I was invited by Istvan Albert, the inventor of the popular Bioinformatics forum Biostars.org, to write a ChIP-seq book chapter for the Biostar Handbook. Please find the PDF in the link. Enjoy!
In Biostar Handbook,
2017
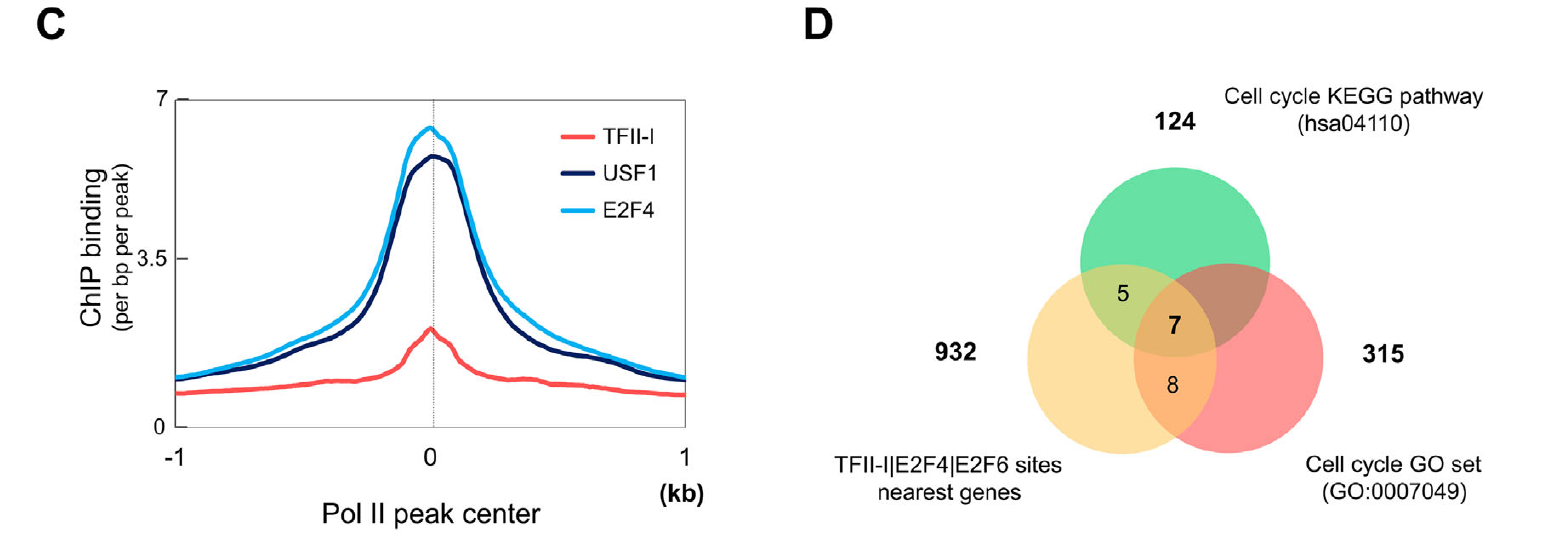
Functional interrelationship between TFII-I and E2F transcription factors at specific cell cycle gene loci
Transcription factor TFII-I is a multifunctional protein implicated in the regulation of cell cycle and stress-response genes. Previous studies have shownthat a subset of TFIII associated genomic sites containedDNA-bindingmotifs for E2F family transcription factors. We analyzed the co-association of TFII-I and E2Fs in more detail using bioinformatics, chromatin immunoprecipitation, and co-immunoprecipitation experiments. The data showthat TFII-I interacts with E2F transcription factors. Furthermore, TFII-I, E2F4, and E2F6 interact with DNA-regulatory elements of several genes implicated in the regulation of the cell cycle, including DNMT1, HDAC1, CDKN1C, and CDC27. Inhibition of TFII-I expression led to a decrease in gene expression and in the association of E2F4 andE2F6 with these gene loci in humanerythroleukemia K562 cells. Finally, TFII-I deficiency reduced the proliferation of K562 cells and increased the sensitivity toward doxorubicin toxicity. The results uncover novel interactions between TFII-I and E2Fs and suggest that TFII-I mediates E2F function at specific cell cycle genes.
In JCB,
2017
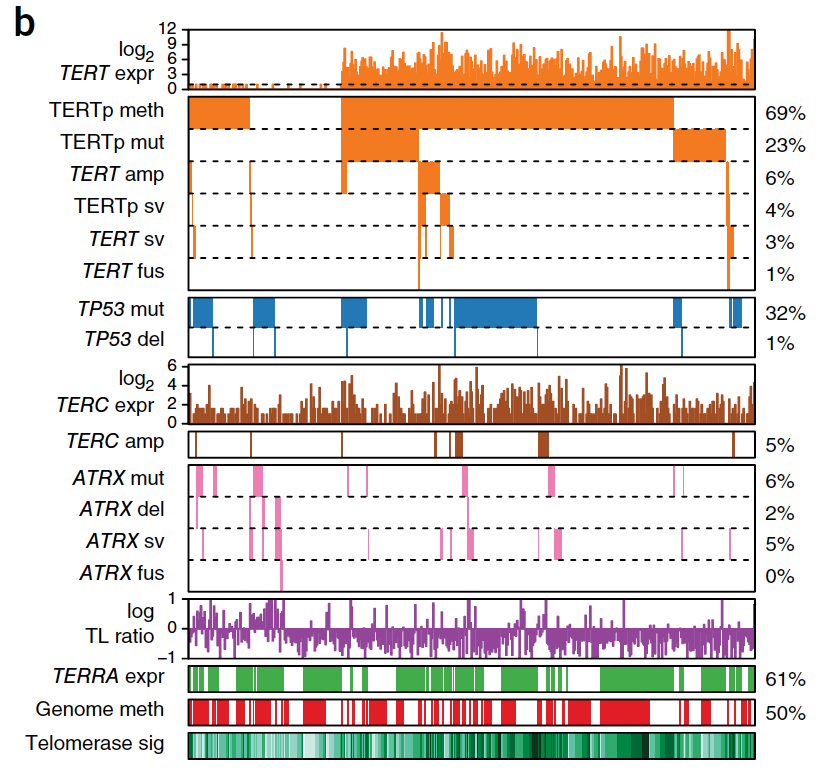
Systematic analysis of telomere length and somatic alterations in 31 cancer types
Cancer cells survive cellular crisis through telomere maintenance mechanisms. We report telomere lengths in 18,430 samples, including tumors and non-neoplastic samples, across 31 cancer types. Telomeres were shorter in tumors than in normal tissues and longer in sarcomas and gliomas than in other cancers. Among 6,835 cancers, 73% expressed telomerase reverse transcriptase (TERT), which was associated with TERT point mutations, rearrangements, DNA amplifications and transcript fusions and predictive of telomerase activity. TERT promoter methylation provided an additional deregulatory TERT expression mechanism. Five percent of cases, characterized by undetectable TERT expression and alterations in ATRX or DAXX, demonstrated elongated telomeres and increased telomeric repeat–containing RNA (TERRA). The remaining 22% of tumors neither expressed TERT nor harbored alterations in ATRX or DAXX. In this group, telomere length positively correlated with TP53 and RB1 mutations. Our analysis integrates TERT abnormalities, telomerase activity and genomic alterations with telomere length in cancer.
In Nature Genetics,
2017
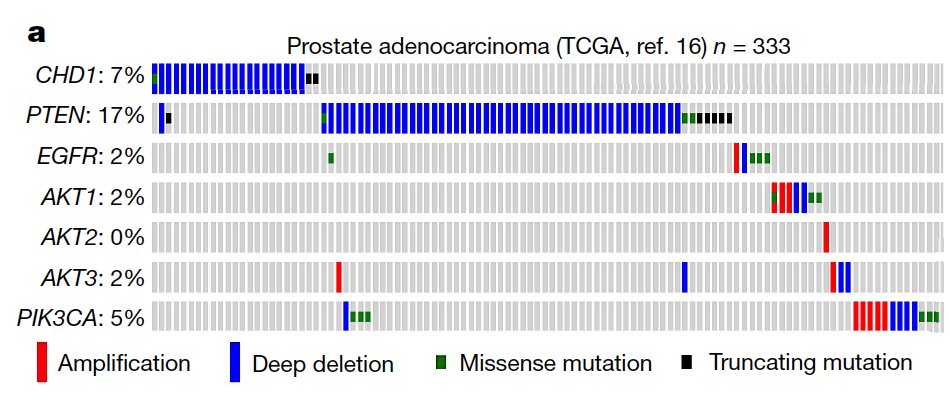
Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer
Synthetic lethality and collateral lethality are two well-validated conceptual strategies for identifying therapeutic targets in cancers with tumour-suppressor gene deletions1–3. Here, we explore an approach to identify potential synthetic-lethal interactions by screening mutually exclusive deletion patterns in cancer genomes. We sought to identify ‘synthetic-essential’ genes: those that are occasionally deleted in some cancers but are almost always retained in the context of a specific tumour-suppressor deficiency. We also posited that such synthetic-essential genes would be therapeutic targets in cancers that harbour specific tumour-suppressor deficiencies. In addition to known synthetic-lethal interactions, this approach uncovered the chromatin helicase DNA-binding factor CHD1 as a putative synthetic-essential gene in PTEN-deficient cancers. In PTEN-deficient prostate and breast cancers, CHD1 depletion profoundly and specifically suppressed cell proliferation, cell survival and tumorigenic potential. Mechanistically, functional PTEN stimulates the GSK3β-mediated phosphorylation of CHD1 degron domains, which promotes CHD1 degradation via the β-TrCP-mediated ubiquitination–proteasome pathway. Conversely, PTEN deficiency results in stabilization of CHD1, which in turn engages the trimethyl lysine-4 histone H3 modification to activate transcription of the pro-tumorigenic TNF–NF-κB gene network. This study identifies a novel PTEN pathway in cancer and provides a framework for the discovery of ‘trackable’ targets in cancers that harbour specific tumour-suppressor deficiencies.
In Nature,
2017
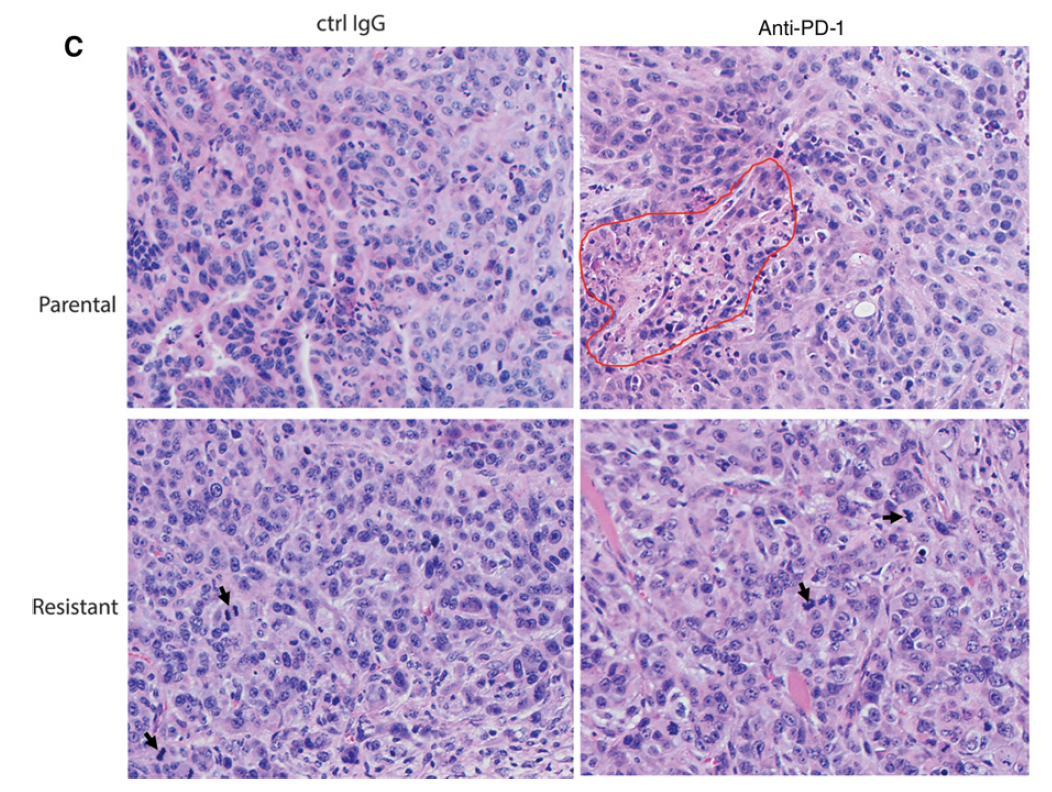
Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy
Immune checkpoint therapies exhibit impressive efficacy in some patients with melanoma or lung cancer, but the lack of response in most cases presses the question of how general efficacy can be improved. In addressing this question, we generated a preclinical tumor model to study anti-PD-1 resistance by in vivo passaging of Kras-mutated, p53-deficient murine lung cancer cells (p53R172HDg/þK-rasLA1/þ) in a syngeneic host exposed to repetitive dosing with anti-mouse PD-1 antibodies. PD-L1 (CD274) expression did not differ between the resistant and parental tumor cells. However, the expression of important molecules in the antigen presentation pathway, including MHC class I and II, as well as b2-microglobulin, were significantly downregulated in the anti-PD-1–resistant tumors compared with parental tumors. Resistant tumors also contained fewer CD8þ (CD8a) and CD4þ tumor-infiltrating lymphocytes and reduced production of IFNg. Localized radiotherapy induced IFNb production, thereby elevating MHC class I expression on both parental and resistant tumor cells and restoring the responsiveness of resistant tumors to anti-PD-1 therapy. Conversely, blockade of type I IFN signaling abolished the effect of radiosensitization in this setting. Collectively, these results identify a mechanism of PD-1 resistance and demonstrate that adjuvant radiotherapy can overcome resistance. These findings have immediate clinical implications for extending the efficacy of anti-PD-1 immune checkpoint therapy in patients.
In Cancer Research,
2017
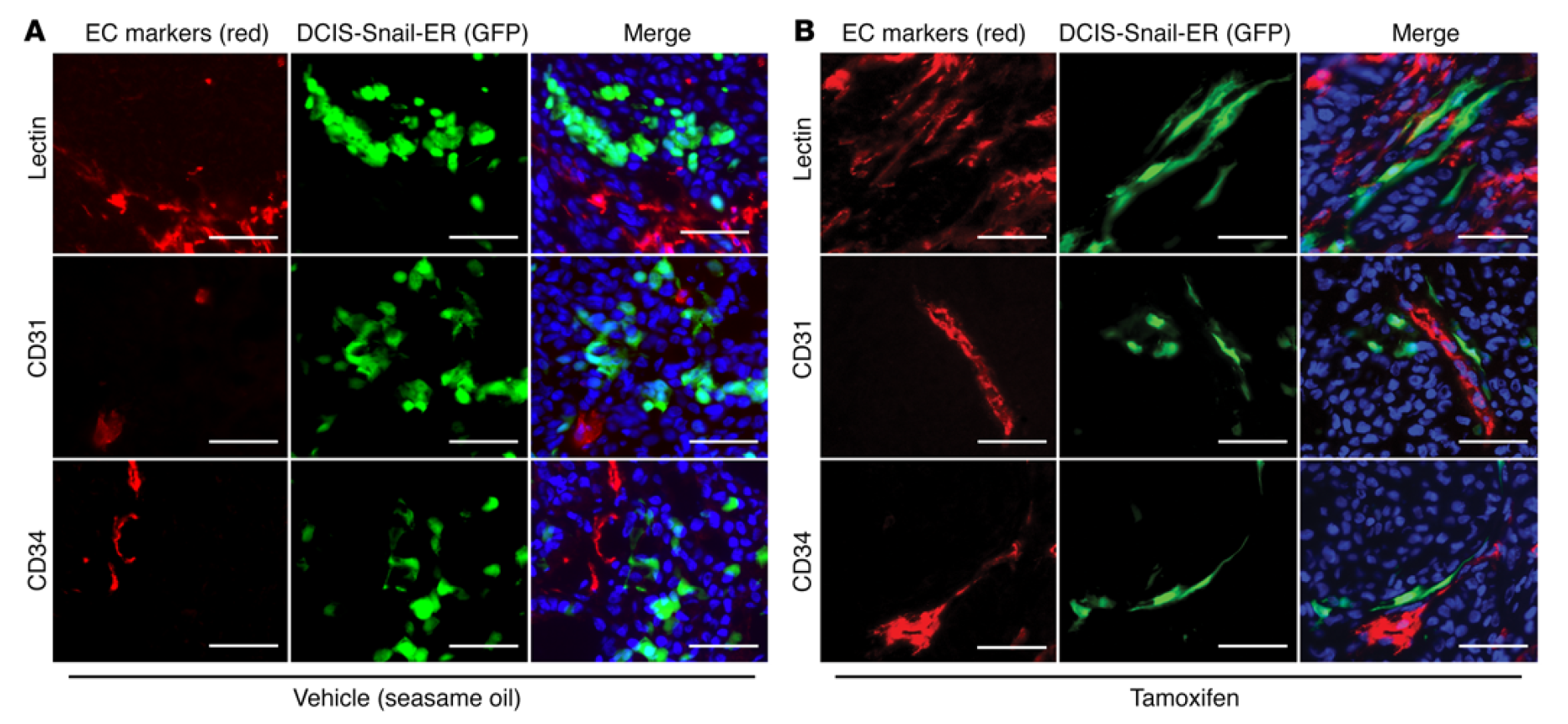
Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells
Carcinoma cells can acquire increased motility and invasiveness through epithelial-to-mesenchymal transition (EMT). However, the significance of EMT in cancer metastasis has been controversial, and the exact fates and functions of EMT cancer cells in vivo remain inadequately understood. Here, we tracked epithelial cancer cells that underwent inducible or spontaneous EMT in various tumor transplantation models. Unlike epithelial cells, the majority of EMT cancer cells were specifically located in the perivascular space and closely associated with blood vessels. EMT markedly activated multiple pericyte markers in carcinoma cells, in particular PDGFR-β and N-cadherin, which enabled EMT cells to be chemoattracted towards and physically interact with endothelium. In tumor xenografts generated from carcinoma cells that were prone to spontaneous EMT, a substantial fraction of the pericytes associated with tumor vasculature were derived from EMT cancer cells. Depletion of such EMT cells in transplanted tumors diminished pericyte coverage, impaired vascular integrity, and attenuated tumor growth. These findings suggest that EMT confers key pericyte attributes on cancer cells. The resulting EMT cells phenotypically and functionally resemble pericytes and are indispensable for vascular stabilization and sustained tumor growth. This study thus proposes a previously unrecognized role for EMT in cancer.
In JCI,
2016
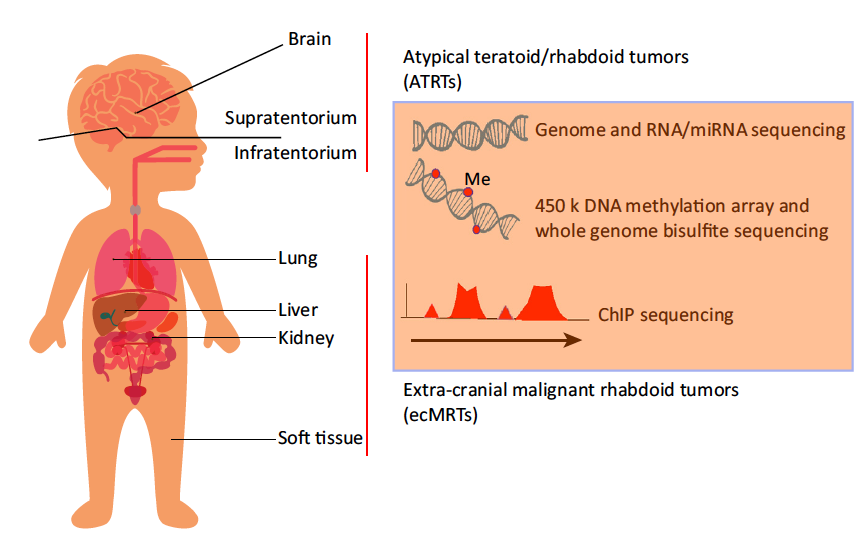
A Molecular Take on Malignant Rhabdoid Tumors
The molecular basis for the clinical heterogeneity observed in patients with malignant rhabdoid tumors is unknown. Recently, two reports revealed molecular intertumor heterogeneity in teratoid/rhabdoid tumors (ATRTs) and extra-cranial MRTs (ecMRTs) using genomic, transcriptomic, and epigenomic profiling. Distinct molecular subgroups were identified and new therapeutic targets were revealed.
In Trends in Cancer,
2016
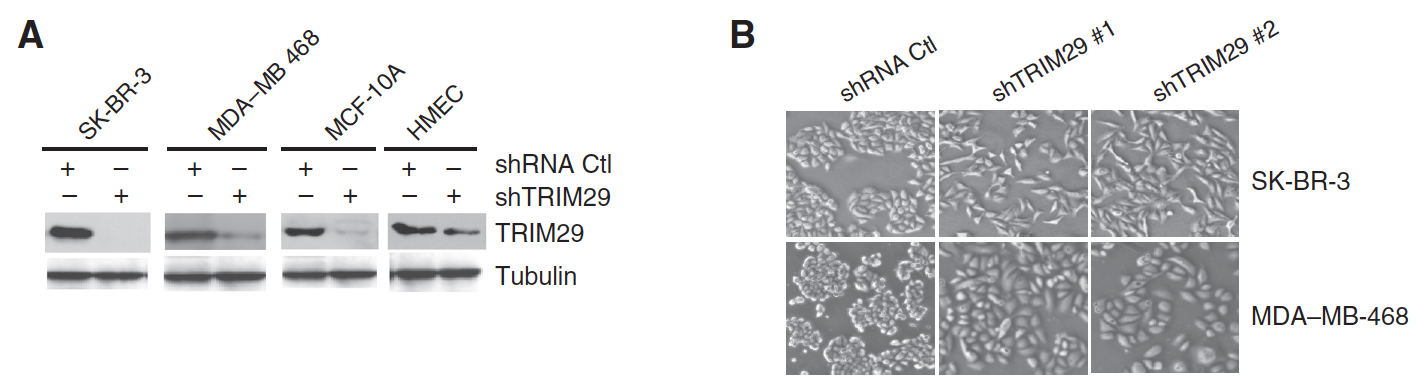
TRIM29 Suppresses TWIST1 and Invasive Breast Cancer Behavior
TRIM29 (ATDC) exhibits a contextual function in cancer, but seems to exert a tumor-suppressor role in breast cancer. Here, we show that TRIM29 is often silenced in primary breast tumors and cultured tumor cells as a result of aberrant gene hypermethylation. RNAi-mediated silencing of TRIM29 in breast tumor cells increased their motility, invasiveness, and proliferation in a manner associated with increased expression of mesenchymal markers (N-cadherin and vimentin), decreased expression of epithelial markers (E-cadherin and EpCAM), and increased expression and activity of the oncogenic transcription factor TWIST1, an important driver of the epithelial–mesenchymal transition (EMT). Functional investigations revealed an inverse relationship in the expression of TRIM29 and TWIST1, suggesting the existence of a negative regulatory feedback loop. In support of this relationship, we found that TWIST1 inhibited TRIM29 promoter activity through direct binding to a region containing a cluster of consensus E-box elements, arguing that TWIST1 transcriptionally represses TRIM29 expression. Analysis of a public breast cancer gene-expression database indicated that reduced TRIM29 expression was associated with reduced relapse-free survival, increased tumor size, grade, and metastatic characteristics. Taken together, our results suggest that TRIM29 acts as a tumor suppressor in breast cancer through its ability to inhibit TWIST1 and suppress EMT.
In Cancer Research,
2014
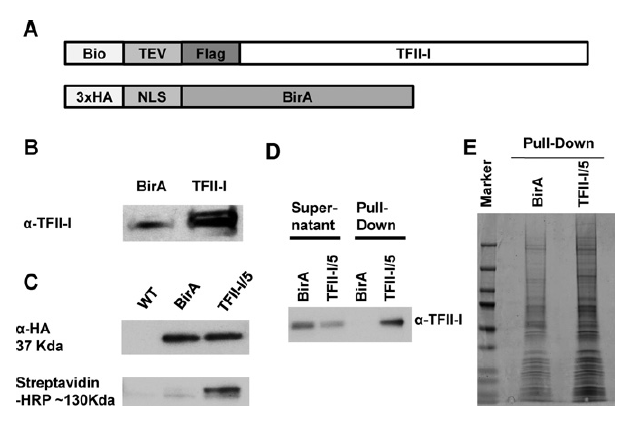
Genomic and proteomic analysis of transcription factor TFII-I reveals insight into the response to cellular stress
The ubiquitously expressed transcription factor TFIII exerts both positive and negative effects on transcription. Using biotinylation tagging technology and high-throughput sequencing, we determined sites of chromatin interactions for TFII-I in the human erythroleukemia cell line K562. This analysis revealed that TFII-I binds upstream of the transcription start site of expressed genes, both upstream and downstream of the transcription start site of repressed genes, and downstream of RNA polymerase II peaks at the ATF3 and other stress responsive genes. At the ATF3 gene, TFII-I binds immediately downstream of a Pol II peak located 5 kb upstream of exon 1. Induction of ATF3 expression increases transcription throughout the ATF3 gene locus which requires TFIII and correlates with increased association of Pol II and Elongin A. Pull-down assays demonstrated that TFII-I interacts with Elongin A. Partial depletion of TFII-I expression caused a reduction in the association of Elongin A with and transcription of the DNMT1 and EFR3A genes without a decrease in Pol II recruitment. The data reveal different interaction patterns of TFII-I at active, repressed, or inducible genes, identify novel TFII-I interacting proteins, implicate TFII-I in the regulation of transcription elongation and provide insight into the role of TFII-I during the response to cellular stress.
In Nucleic Acids Research,
2014
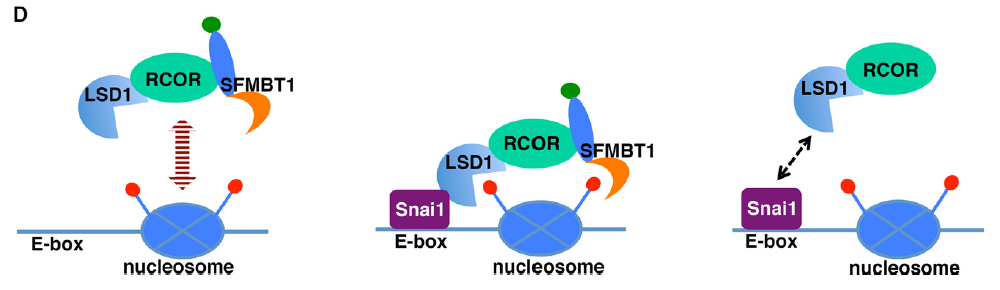
The malignant brain tumor (MBT) domain protein SFMBT1 is an integral histone reader subunit of the LSD1 demethylase complex for chromatin association and epithelial-to-mesenchymal transition
Chromatin readers decipher the functional readouts of histone modifications by recruiting specific effector complexes for subsequent epigenetic reprogramming. The LSD1 (also known as KDM1A) histone demethylase complex modifies chromatin and represses transcription in part by catalyzing demethylation of dimethylated histone H3 lysine 4 (H3K4me2), a mark for active transcription. However, none of its currently known subunits recognizes methylated histones. The Snai1 family transcription factors are central drivers of epithelial-to-mesenchymal transition (EMT) by which epithelial cells acquire enhanced invasiveness. Snai1-mediated transcriptional repression of epithelial genes depends on its recruitment of the LSD1 complex and ensuing demethylation of H3K4me2 at its target genes. Through biochemical purification, we identified the MBT domain-containing protein SFMBT1 as a novel component of the LSD1 complex associated with Snai1. Unlike other mammalian MBT domain proteins characterized to date that selectively recognize mono- and dimethylated lysines, SFMBT1 binds diand trimethyl H3K4, both of which are enriched at active promoters. We show that SFMBT1 is essential for Snai1-dependent recruitment of LSD1 to chromatin, demethylation of H3K4me2, transcriptional repression of epithelial markers, and induction of EMT by TGFb. Carcinogenic metal nickel is a widespread environmental and occupational pollutant. Nickel alters gene expression and induces EMT. We demonstrate the nickel-initiated effects are dependent on LSD1-SFMBT1-mediated chromatin modification. Furthermore, in human cancer, expression of SFMBT1 is associated with mesenchymal markers and unfavorable prognosis. These results highlight a critical role of SFMBT1 in epigenetic regulation, EMT, and cancer.
In JBC,
2013
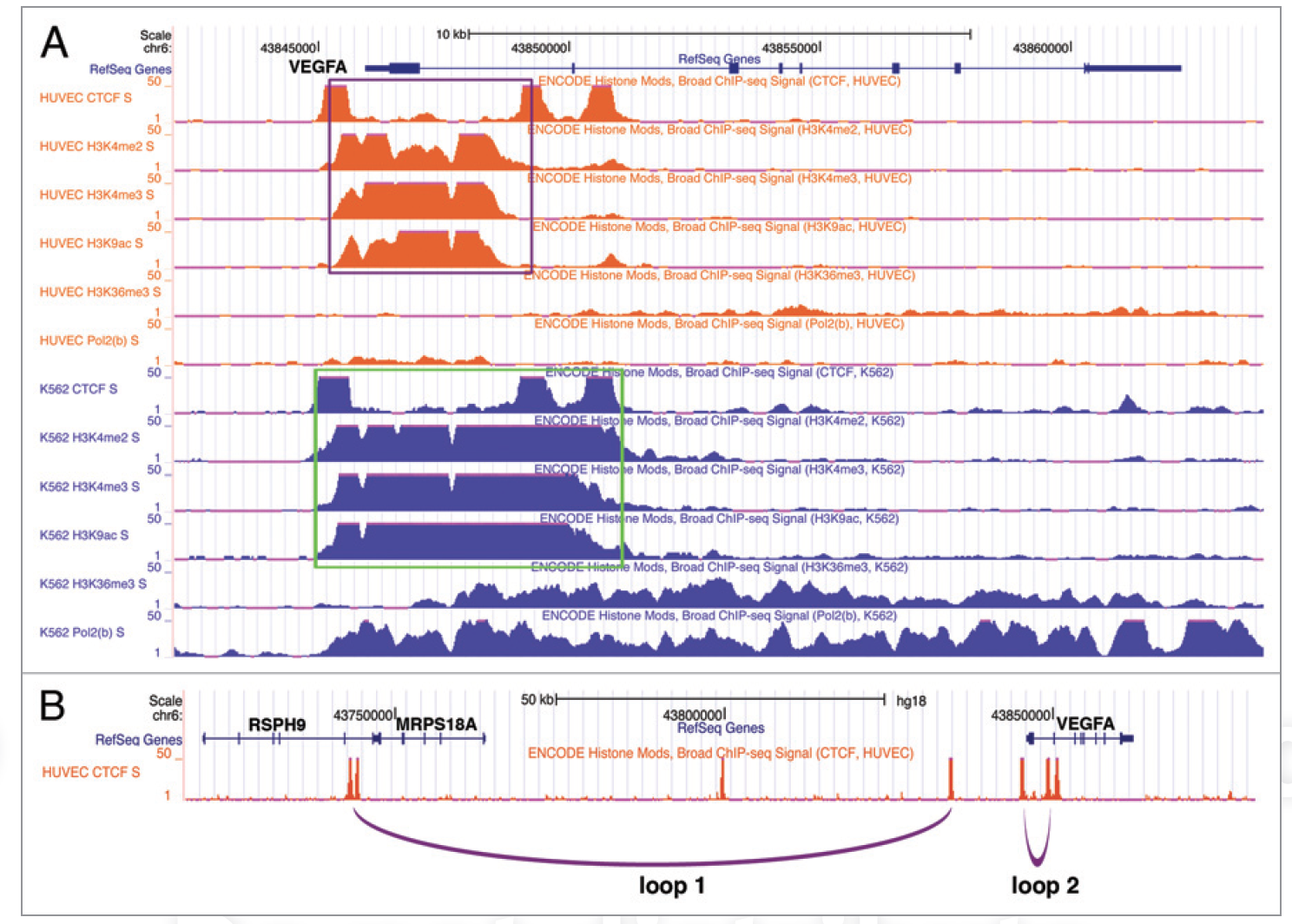
CTCF-dependent chromatin insulator as a built-in attenuator of angiogenesis
VEGF is a pivotal pro-angiogenic growth factor and its dosage decisively impacts vascularization. We recently identified a CTCF-dependent chromatin insulator that critically restrains the transcriptional induction of VEGF and angiogenesis. We postulate that CTCF may exert enhancer blocking by mediating chromatin looping and/or RNA polymerase pausing at the VEGF locus.
In Transcription,
2012
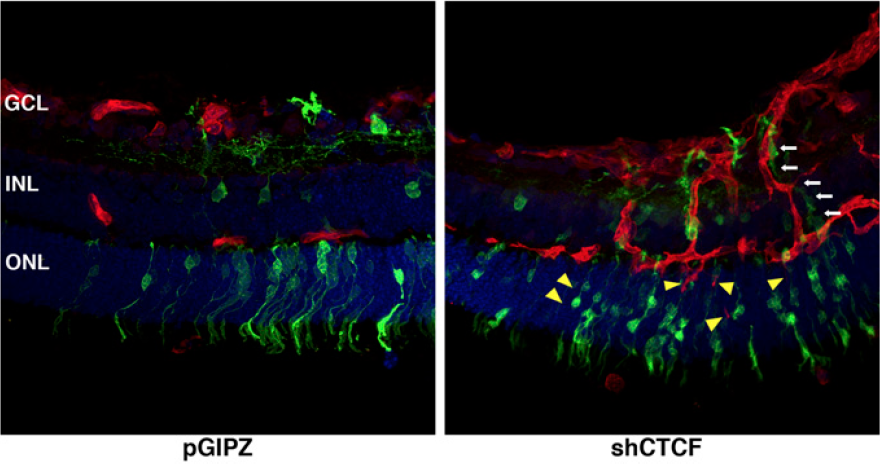
Restraint of angiogenesis by zinc finger transcription factor CTCF-dependent chromatin insulation
Angiogenesis is meticulously controlled by a fine balance between positive and negative regulatory activities. Vascular endothelial growth factor (VEGF) is a predominant angiogenic factor and its dosage is precisely regulated during normal vascular formation. In cancer, VEGF is commonly overproduced, resulting in abnormal neovascularization. VEGF is induced in response to various stimuli including hypoxia; however, very little is known about the mechanisms that confine its induction to ensure proper angiogenesis. Chromatin insulation is a key transcription mechanism that prevents promiscuous gene activation by interfering with the action of enhancers. Here we show that the chromatin insulator-binding factor CTCF binds to the proximal promoter of VEGF. Consistent with the enhancer-blocking mode of chromatin insulators, CTCF has little effect on basal expression of VEGF but specifically affects its activation by enhancers. CTCF knockdown cells are sensitized for induction of VEGF and exhibit elevated proangiogenic potential. Cancer-derived CTCF missense mutants are mostly defective in blocking enhancers at the VEGF locus. Moreover, during mouse retinal development, depletion of CTCF causes excess angiogenesis. Therefore, CTCF-mediated chromatin insulation acts as a crucial safeguard against hyperactivation of angiogenesis.
In PNAS,
2011