 My caption 😄
My caption 😄
CHIPS: A Snakemake pipeline for quality control and reproducible processing of chromatin profiling data
Abstract
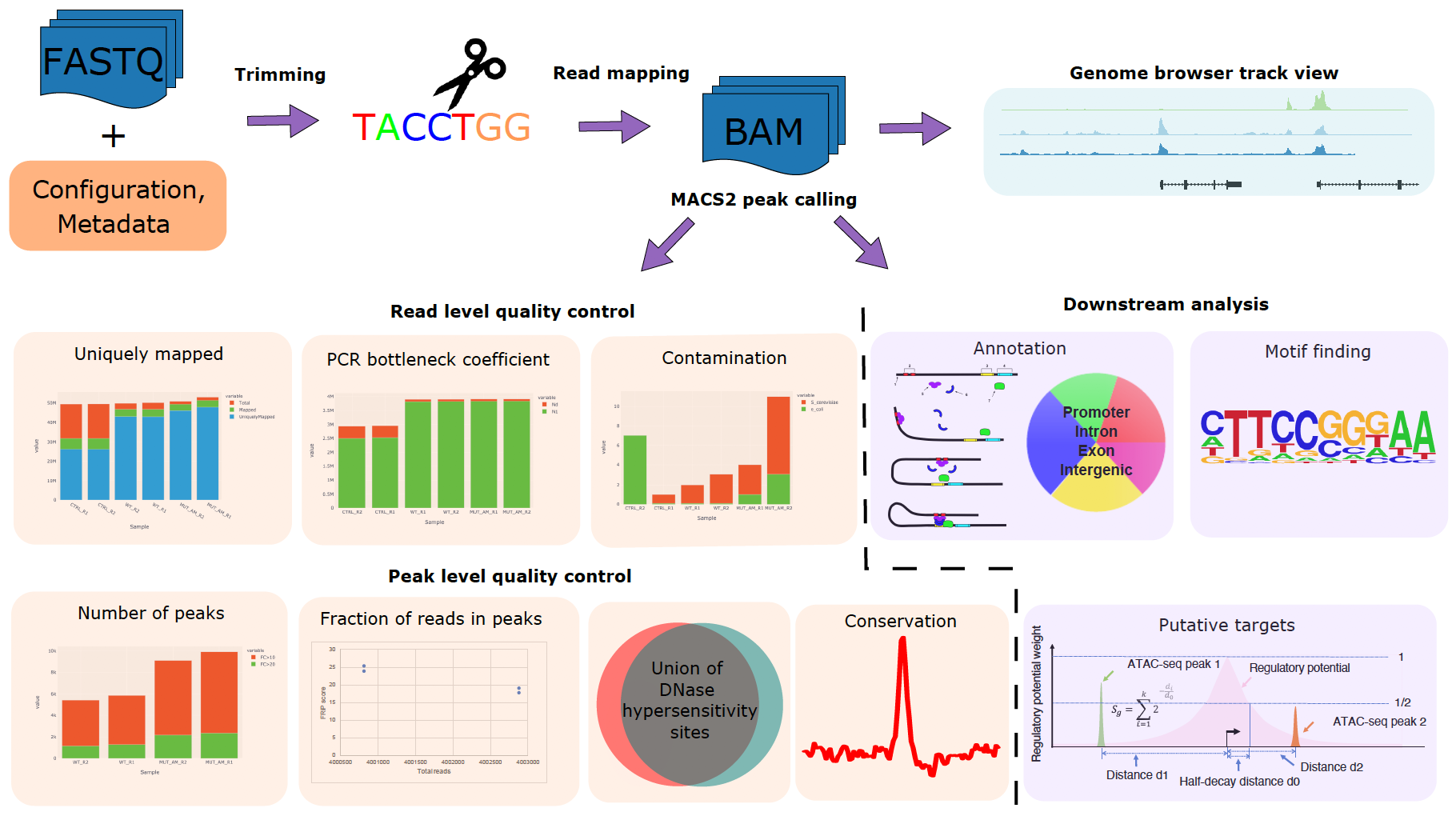
Motivation: The chromatin profile measured by ATAC-seq, ChIP-seq, or DNase-seq experiments can identify genomic regions critical in regulating gene expression and provide insights on biological processes such as diseases and development. However, quality control and processing chromatin profiling data involve many steps, and different bioinformatics tools are used at each step. It can be challenging to manage the analysis. Results: We developed a Snakemake pipeline called CHIPS (CHromatin enrichment Processor) to streamline the processing of ChIP-seq, ATAC-seq, and DNase-seq data. The pipeline supports single- and paired-end data and is flexible to start with FASTQ or BAM files. It includes basic steps such as read trimming, mapping, and peak calling. In addition, it calculates quality control metrics such as contamination profiles, PCR bottleneck coefficient, the fraction of reads in peaks, percentage of peaks overlapping with the union of public DNaseI hypersensitivity sites, and conservation profile of the peaks. For downstream analysis, it carries out peak annotations, motif finding, and regulatory potential calculation for all genes. The pipeline ensures that the processing is robust and reproducible. Availability: CHIPS is available at https://bitbucket.org/plumbers/cidc_chips/src/master/ Contact: mtang@ds.dfci.harvard.edu; henry_long@dfci.harvard.edu
More detail can easily be written here using Markdown and $\rm \LaTeX$ math code.