To not miss a post like this, sign up for my newsletter to learn computational biology and bioinformatics.
In my last post, I showed you how to use PCA for bulk RNAseq data.
Today, let’s see how we can use it for scATACseq data.
Download the example dataset from 10x genomics https://support.10xgenomics.com/single-cell-atac/datasets/1.1.0/atac_pbmc_5k_v1
The dataset is 5k Peripheral blood mononuclear cells (PBMCs) from a healthy donor (v1.0).
Download the atac_pbmc_5k_v1_filtered_peak_bc_matrix.tar.gz file and unzip it.
It will give you a folder filtered_peak_bc_matrix containing three files:
- barcodes.tsv
- matrix.mtx
- peaks.bed
library(Seurat)
library(Matrix)
library(readr)
library(dplyr)
mat<- readMM("~/blog_data/filtered_peak_bc_matrix/matrix.mtx")
# this is a sparse matrix
mat[1:5, 1:5]#> 5 x 5 sparse Matrix of class "dgTMatrix"
#>
#> [1,] . . . . .
#> [2,] . . . . .
#> [3,] . . . . .
#> [4,] . . . . .
#> [5,] . . 2 2 .peaks<- read_tsv("~/blog_data/filtered_peak_bc_matrix/peaks.bed", col_names = FALSE)
head(peaks)#> # A tibble: 6 × 3
#> X1 X2 X3
#> <chr> <dbl> <dbl>
#> 1 chr1 569196 569616
#> 2 chr1 713593 714659
#> 3 chr1 752507 752955
#> 4 chr1 762419 763295
#> 5 chr1 804991 805709
#> 6 chr1 839797 840795peaks<- peaks %>%
mutate(peak = paste(X1,X2,X3, sep="_"))
barcodes<- read_tsv("~/blog_data/filtered_peak_bc_matrix/barcodes.tsv", col_names = FALSE)
head(barcodes)#> # A tibble: 6 × 1
#> X1
#> <chr>
#> 1 AAACGAAAGACACTTC-1
#> 2 AAACGAAAGCATACCT-1
#> 3 AAACGAAAGCGCGTTC-1
#> 4 AAACGAAAGGAAGACA-1
#> 5 AAACGAACAGGCATCC-1
#> 6 AAACGAAGTTTGTCTT-1set the rownames and colnames for the count matrix
rownames(mat)<- peaks$peak
colnames(mat)<- barcodes$X1
mat[1:5, 1:5]#> 5 x 5 sparse Matrix of class "dgTMatrix"
#> AAACGAAAGACACTTC-1 AAACGAAAGCATACCT-1 AAACGAAAGCGCGTTC-1
#> chr1_569196_569616 . . .
#> chr1_713593_714659 . . .
#> chr1_752507_752955 . . .
#> chr1_762419_763295 . . .
#> chr1_804991_805709 . . 2
#> AAACGAAAGGAAGACA-1 AAACGAACAGGCATCC-1
#> chr1_569196_569616 . .
#> chr1_713593_714659 . .
#> chr1_752507_752955 . .
#> chr1_762419_763295 . .
#> chr1_804991_805709 2 .check the range of the matrix
range(mat)#> [1] 0 62dim(mat)#> [1] 47520 4621It has 47520 peaks and 4621 cells.
PCA for the raw counts
Let’s calculate the PCA from scratch with irlba for big matrix. Use built-in svd if the matrix is small or use prcomp. They are all for the same purpose: calculate PCA.
mat.pca<- irlba::irlba(t(mat))
cell_embeddings<- mat.pca$u %*% diag(mat.pca$d)
dim(cell_embeddings)#> [1] 4621 5cell_embeddings[1:5, 1:5]#> [,1] [,2] [,3] [,4] [,5]
#> [1,] 80.37159 -7.069846 -6.9684003 -4.4335265 -4.60719405
#> [2,] 109.67981 36.289439 0.1593661 1.8245021 -0.24826248
#> [3,] 55.16413 19.974192 0.2210001 -3.7452100 -4.96653834
#> [4,] 71.95250 25.298451 2.3182169 -0.5619408 3.07409882
#> [5,] 77.84515 27.053327 -0.2662783 -1.8013677 0.06394454plot(cell_embeddings[, 1], cell_embeddings[,2])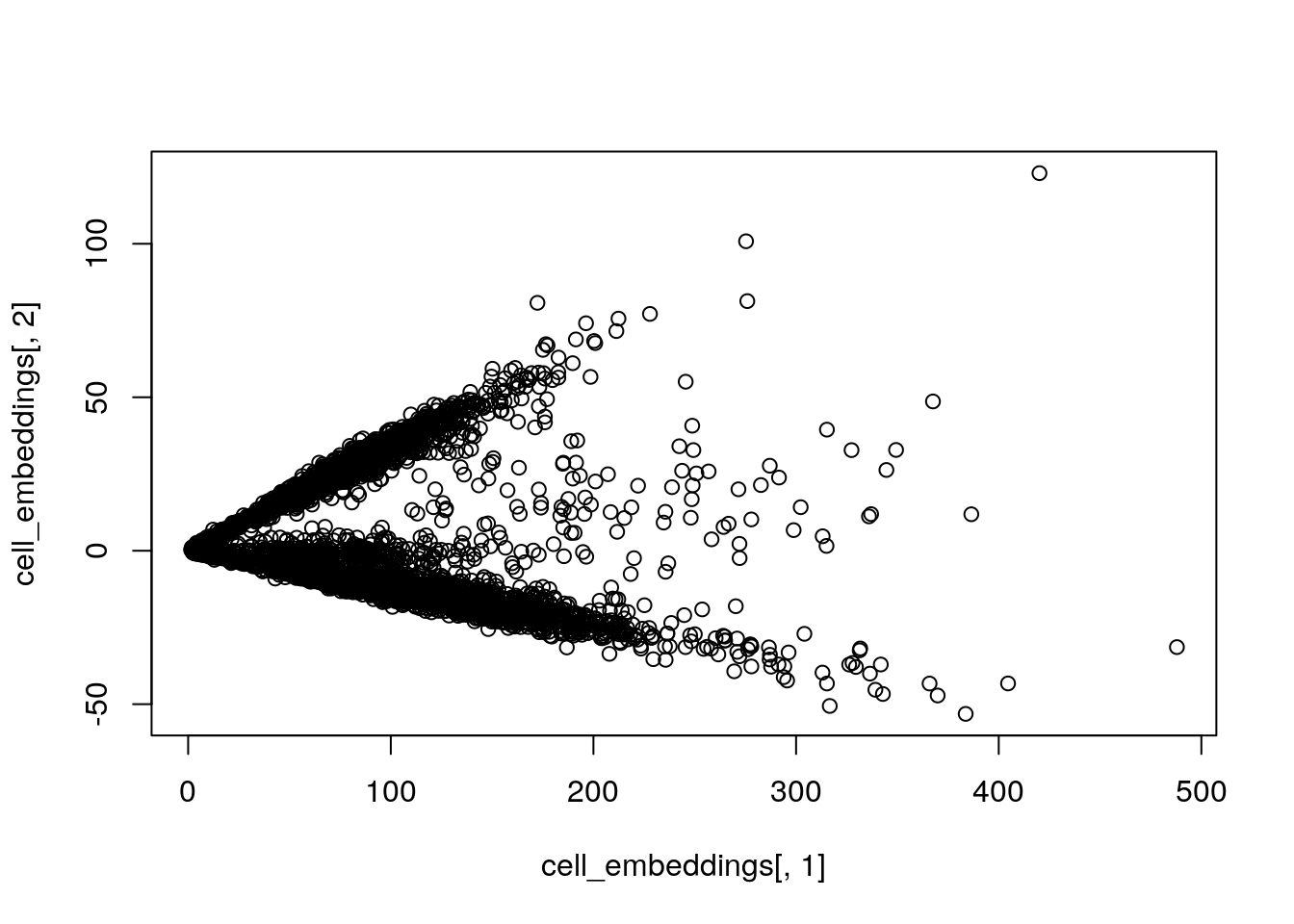
The PCA looks a little strange.
PCA after TF-IDF transformation
Latent Semantic Indexing or LSI method and used a transformation of the binarized scATAC count matrix called ’TF-IDF` (term frequency–inverse document frequency) which is used in text mining followed by PCA.
What is TF-IDF Transformation and Why is it Useful in scATAC-seq Analysis?
TF-IDF (Term Frequency-Inverse Document Frequency) is a numerical transformation originally from text mining. In scATAC-seq, it helps normalize sparse and binary data to highlight important peaks for downstream analysis.
In text mining, TF-IDF measures how important a word (term) is in a document relative to all other documents. It consists of:
🔹 TF (Term Frequency) → How often a term appears in a document.
🔹 IDF (Inverse Document Frequency) → Downweights commonly occurring terms across all documents.
Mathematically:
\[
TF = \frac{\text{Raw count of term in a document}}{\text{Total terms in the document}}
\]
\[
IDF = \log \left( \frac{\text{Total number of documents}}{\text{Number of documents containing the term}} \right)
\]
\[
TF-IDF = TF \times IDF
\]
Why is TF-IDF Used in scATAC-seq?
Unlike scRNA-seq (which has more continuous expression values), scATAC-seq data is largely binary (1 = accessible, 0 = inaccessible). Raw counts are highly sparse and noisy.
Problem: Some peaks (e.g., promoters of housekeeping genes) are open in many cells and dominate the dataset.
Solution: TF-IDF downweights ubiquitous peaks and emphasizes cell-type-specific regulatory regions.
Let’s implement it in R:
# make a copy of the original matrix
mat2<- mat
# binarize the matrix
mat2@x[mat2@x >0]<- 1
# a custom function to perform TF-IDF transformation
TF.IDF.custom <- function(data, verbose = TRUE) {
if (class(x = data) == "data.frame") {
data <- as.matrix(x = data)
}
if (class(x = data) != "dgCMatrix") {
data <- as(object = data, Class = "dgCMatrix")
}
if (verbose) {
message("Performing TF-IDF normalization")
}
npeaks <- Matrix::colSums(x = data)
tf <- t(x = t(x = data) / npeaks)
# log transformation
idf <- log(1+ ncol(x = data) / Matrix::rowSums(x = data))
norm.data <- Diagonal(n = length(x = idf), x = idf) %*% tf
norm.data[which(x = is.na(x = norm.data))] <- 0
return(norm.data)
}PCA on the TF-IDF transformed matrix
mat2<- TF.IDF.custom(mat2)
# what's the range after transformation?
range(mat2)#> [1] 0.00000000 0.04444816mat2[1:5, 1:5]#> 5 x 5 sparse Matrix of class "dgCMatrix"
#> AAACGAAAGACACTTC-1 AAACGAAAGCATACCT-1 AAACGAAAGCGCGTTC-1
#> [1,] . . .
#> [2,] . . .
#> [3,] . . .
#> [4,] . . .
#> [5,] . . 0.0006933661
#> AAACGAAAGGAAGACA-1 AAACGAACAGGCATCC-1
#> [1,] . .
#> [2,] . .
#> [3,] . .
#> [4,] . .
#> [5,] 0.0005400022 .library(irlba)
set.seed(123)
mat.lsi<- irlba::irlba(t(mat2), 50)
# peak_loadings <- mat.lsi$v
cell_embeddings <- mat.lsi$u %*% diag(mat.lsi$d) # Cell embeddings (5k cells in rows)
dim(cell_embeddings)#> [1] 4621 50cell_embeddings[1:5, 1:5]#> [,1] [,2] [,3] [,4] [,5]
#> [1,] -0.01188588 -0.003586754 2.441022e-05 0.0001228626 1.196982e-04
#> [2,] -0.01110892 0.005137220 1.483881e-04 -0.0002180946 -1.504167e-04
#> [3,] -0.01133256 0.005288389 8.627158e-05 -0.0001686069 -3.212530e-04
#> [4,] -0.01113891 0.005190526 1.798458e-04 -0.0005039129 -7.432966e-05
#> [5,] -0.01108726 0.005486348 1.156304e-04 -0.0001435541 -5.605533e-05plot(cell_embeddings[, 1], cell_embeddings[, 2])
You can see that PCA on the TF-IDF transformed matrix reveals more distinct clusters.
Conclusions
LSI is just PCA on the binarized scATACseq count matrix after TF-IDF transformed.
In scATAC-seq, TF-IDF transformation helps normalize sparse and binary data to highlight important peaks for downstream analysis.
Read my old blog post clustering scATACseq data: the TF-IDF way
Bonus: How to read PCA plots